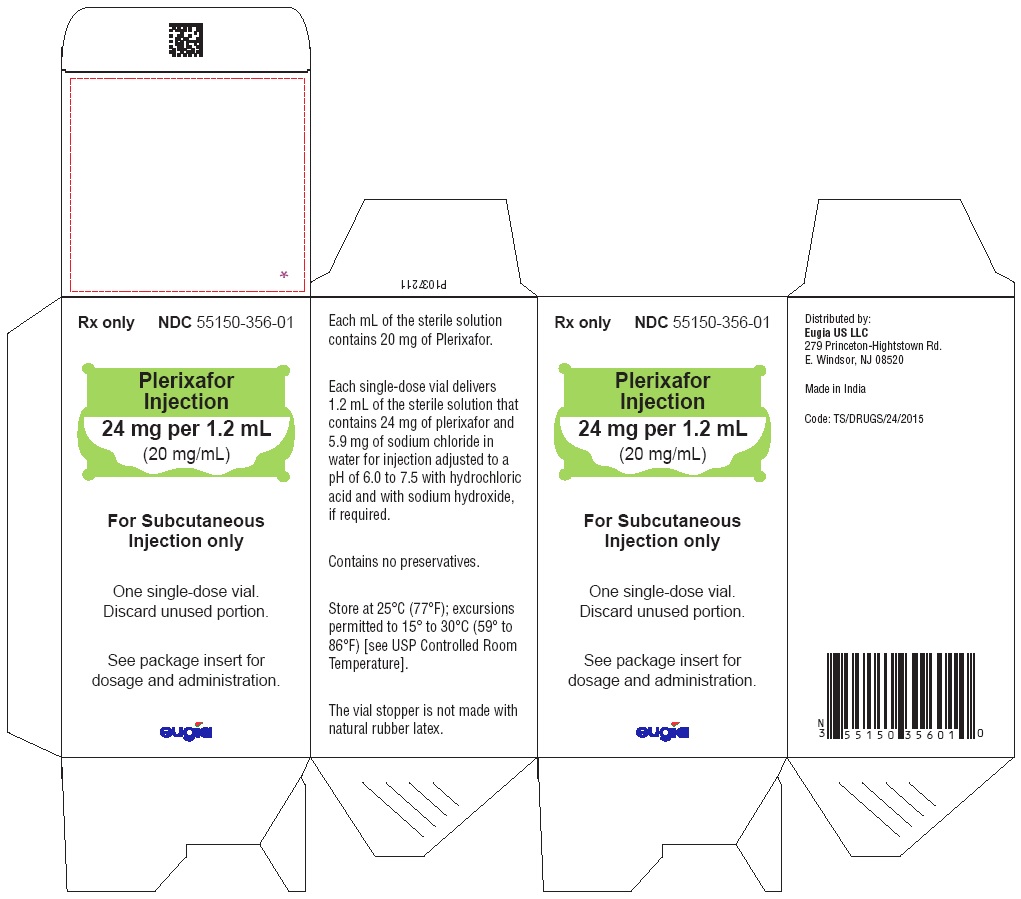
Label: PLERIXAFOR solution


- NDC Code(s): 55150-356-01
- Packager: Eugia US LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated June 28, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PLERIXAFOR INJECTION safely and effectively. See full prescribing information for PLERIXAFOR INJECTION.
PLERIXAFOR injection, for subcutaneous use
Initial U.S. Approval: 2008INDICATIONS AND USAGE
Plerixafor injection, a hematopoietic stem cell mobilizer, is indicated in combination with filgrastim to mobilize hematopoietic stem cells (HSCs) to the peripheral blood for collection and subsequent autologous transplantation in patients with non-Hodgkin’s lymphoma or multiple myeloma. (1)
DOSAGE AND ADMINISTRATION
- Initiate plerixafor injection treatment after the patient has received filgrastim once daily for 4 days. (2.1)
- Repeat plerixafor injection dose up to 4 consecutive days. (2.1)
- Dose based on patient weight
- Administer by subcutaneous injection approximately 11 hours prior to initiation of apheresis. (2.1)
- Renal impairment: If creatinine clearance is ≤50 mL/min, decrease dose by one-third to 0.16 mg/kg. (2.3)
DOSAGE FORMS AND STRENGTHS
- Injection: 24 mg/1.2 mL (20 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
- History of hypersensitivity to plerixafor. (4)
WARNINGS AND PRECAUTIONS
- Anaphylactic Shock and Serious Hypersensitivity Reactions have occurred. Monitor patients during and after completion of plerixafor administration. (5.1)
- Tumor Cell Mobilization in Leukemia Patients: plerixafor may mobilize leukemic cells and should not be used in leukemia patients. (5.2)
- Hematologic Effects: Increased circulating leukocytes and decreased platelet counts have been observed. Monitor blood cell counts and platelet counts during plerixafor use. (5.3)
- Potential for Tumor Cell Mobilization: Tumor cells may be released from marrow during HSC mobilization with plerixafor and filgrastim. Effect of reinfusion of tumor cells is unknown. (5.4)
- Splenic Rupture: Evaluate patients who report left upper abdominal and/or scapular or shoulder pain. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise women not to become pregnant when taking plerixafor. (5.6, 8.1)
ADVERSE REACTIONS
Most common adverse reactions (≥10%): diarrhea, nausea, fatigue, injection site reactions, headache, arthralgia, dizziness, and vomiting. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Eugia US LLC at 1-866-850-2876 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Administration
2.2 Recommended Concomitant Medications
2.3 Dose Modifications in Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactic Shock and Hypersensitivity Reactions
5.2 Tumor Cell Mobilization in Leukemia Patients
5.3 Hematologic Effects
5.4 Potential for Tumor Cell Mobilization
5.5 Splenic Enlargement and Rupture
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Administration
Begin treatment with plerixafor injection after the patient has received filgrastim once daily for 4 days [see Dosage and Administration (2.2)]. Administer plerixafor injection approximately 11 hours prior to initiation of each apheresis for up to 4 consecutive days.
The recommended dose of plerixafor injection by subcutaneous injection is based on body weight:- 20 mg fixed dose or 0.24 mg/kg of body weight for patients weighing less than or equal to 83 kg. [see Clinical Pharmacology (12.3)]
- 0.24 mg/kg of body weight for patients weighing greater than 83 kg
Use the patient’s actual body weight to calculate the volume of plerixafor injection to be administered. Each vial delivers 1.2 mL of 20 mg/mL solution, and the volume to be administered to patients should be calculated from the following equation:
0.012 × patient’s actual body weight (in kg) = volume to be administered (in mL)
In clinical studies, plerixafor injection dose has been calculated based on actual body weight in patients up to 175% of ideal body weight. Plerixafor injection dose and treatment of patients weighing more than 175% of ideal body weight have not been investigated.
Based on increasing exposure with increasing body weight, the plerixafor injection dose should not exceed 40 mg/day [see Clinical Pharmacology (12.3)].
Vials should be inspected visually for particulate matter and discoloration prior to administration and should not be used if there is particulate matter or if the solution is discolored.
Discard unused portion.2.2 Recommended Concomitant Medications
Administer daily morning doses of filgrastim 10 mcg/kg for 4 days prior to the first evening dose of plerixafor injection and on each day prior to apheresis [see Clinical Studies (14)].
2.3 Dose Modifications in Renal Impairment
In patients with moderate and severe renal impairment (estimated creatinine clearance (CLCR) less than or equal to 50 mL/min), reduce the dose of plerixafor injection by one-third based on body weight category as shown in Table 1. If CLCR is less than or equal to 50 mL/min the dose should not exceed 27 mg/day, as the mg/kg-based dosage results in increased plerixafor exposure with increasing body weight [see Clinical Pharmacology (12.3)]. Similar systemic exposure is predicted if the dose is reduced by one-third in patients with moderate and severe renal impairment compared with subjects with normal renal function [see Clinical Pharmacology (12.3)].
Table 1: Recommended Dosage of Plerixafor Injection in Patients with Renal Impairment Estimated Creatinine Clearance (mL/min)
Dose
Body Weight less than or equal to 83 kg
Body Weight greater than 83 kg
and less than 160 kg
greater than 50
20 mg or 0.24 mg/kg once daily
0.24 mg/kg once daily
(not to exceed 40 mg/day)
less than or equal to 50
13 mg or 0.16 mg/kg once daily
0.16 mg/kg once daily
(not to exceed 27 mg/day)
The following (Cockcroft-Gault) formula may be used to estimate CLCR:
Males:
Creatinine clearance (mL/min) = weight (kg) × (140 – age in years)
72 × serum creatinine (mg/dL)
Females:
Creatinine clearance (mL/min) = 0.85 × value calculated for males
There is insufficient information to make dosage recommendations in patients on hemodialysis.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Plerixafor injection is contraindicated in patients with a history of hypersensitivity to plerixafor [see Warnings and Precautions (5.1)]. Anaphylactic shock has occurred with use of plerixafor.
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactic Shock and Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening with clinically significant hypotension and shock have occurred in patients receiving plerixafor [see Adverse Reactions (6.2)]. Observe patients for signs and symptoms of hypersensitivity during and after plerixafor administration for at least 30 minutes and until clinically stable following completion of each administration. Only administer plerixafor when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
In clinical studies, mild or moderate allergic reactions occurred within approximately 30 minutes after plerixafor administration in less than 1% of patients [see Adverse Reactions (6.1)].5.2 Tumor Cell Mobilization in Leukemia Patients
For the purpose of HSC mobilization, plerixafor may cause mobilization of leukemic cells and subsequent contamination of the apheresis product. Therefore, plerixafor is not intended for HSC mobilization and harvest in patients with leukemia.
5.3 Hematologic Effects
Leukocytosis
Administration of plerixafor in conjunction with filgrastim increases circulating leukocytes as well as HSC populations. Monitor white blood cell counts during plerixafor use [see Adverse Reactions (6.1)].
Thrombocytopenia
Thrombocytopenia has been observed in patients receiving plerixafor. Monitor platelet counts in all patients who receive plerixafor and then undergo apheresis.5.4 Potential for Tumor Cell Mobilization
When plerixafor is used in combination with filgrastim for HSC mobilization‚ tumor cells may be released from the marrow and subsequently collected in the leukapheresis product. The effect of potential reinfusion of tumor cells has not been well-studied.
5.5 Splenic Enlargement and Rupture
Higher absolute and relative spleen weights associated with extramedullary hematopoiesis were observed following prolonged (2 to 4 weeks) daily plerixafor subcutaneous administration in rats at doses approximately 4-fold higher than the recommended human dose based on body surface area. The effect of plerixafor on spleen size in patients was not specifically evaluated in clinical studies. Cases of splenic enlargement and/or rupture have been reported following the administration of plerixafor in conjunction with filgrastim. Evaluate individuals receiving plerixafor in combination with filgrastim who report left upper abdominal pain and/or scapular or shoulder pain for splenic integrity.
5.6 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, plerixafor can cause fetal harm when administered to a pregnant woman. Plerixafor administration to pregnant rats during organogenesis resulted in embryo-fetal mortality, structural abnormalities, and alterations to growth at exposures approximately 10 times the exposure at the recommended human dose.
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use an effective form of contraception during treatment with plerixafor and for one week after the final dose [see Use in Specific Populations (8.1)]. -
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Anaphylactic shock and hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Potential for tumor cell mobilization in leukemia patients [see Warnings and Precautions (5.2)]
- Increased circulating leukocytes and decreased platelet counts [see Warnings and Precautions (5.3)]
- Potential for tumor cell mobilization [see Warnings and Precautions (5.4)]
- Splenic enlargement [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions (≥10%) reported in patients who received plerixafor in conjunction with filgrastim regardless of causality and more frequent with plerixafor than placebo during HSC mobilization and apheresis were diarrhea, nausea, fatigue, injection site reactions, headache, arthralgia, dizziness, and vomiting.
Safety data for plerixafor in combination with filgrastim were obtained from two randomized placebo-controlled studies (301 patients) and 10 uncontrolled studies (242 patients). Patients were primarily treated with plerixafor at daily doses of 0.24 mg/kg subcutaneous. Median exposure to plerixafor in these studies was 2 days (range 1 to 7 days).
In the two randomized studies in patients with NHL and MM, a total of 301 patients were treated in the plerixafor and filgrastim group and 292 patients were treated in the placebo and filgrastim group. Patients received daily morning doses of filgrastim 10 mcg/kg for 4 days prior to the first dose of plerixafor 0.24 mg/kg subcutaneous or placebo and on each morning prior to apheresis. The adverse reactions that occurred in ≥5% of the patients who received plerixafor regardless of causality and were more frequent with plerixafor than placebo during HSC mobilization and apheresis are shown in Table 2.
Table 2: Adverse Reactions in ≥5% of Non-Hodgkin’s Lymphoma and Multiple Myeloma Patients Receiving Plerixafor and More Frequent than Placebo during HSC Mobilization and Apheresis * Grades based on criteria from the World Health Organization (WHO)
Percent of Patients (%)
Plerixafor and Filgrastim
(n=301)
Placebo and Filgrastim
(n=292)
All Grades*
Grade
3
Grade
4
All
Grades
Grade
3
Grade
4
Gastrointestinal disorders
Diarrhea
37
<1
0
17
0
0
Nausea
34
1
0
22
0
0
Vomiting
10
<1
0
6
0
0
Flatulence
7
0
0
3
0
0
General disorders and
administration site conditions
Injection site reactions
34
0
0
10
0
0
Fatigue
27
0
0
25
0
0
Musculoskeletal and connective
tissue disorders
Arthralgia
13
0
0
12
0
0
Nervous system disorders
Headache
22
<1
0
21
1
0
Dizziness
11
0
0
6
0
0
Psychiatric disorders
Insomnia
7
0
0
5
0
0
In the randomized studies, 34% of patients with NHL or MM had mild to moderate injection site reactions at the site of subcutaneous administration of plerixafor. These included erythema, hematoma, hemorrhage, induration, inflammation, irritation, pain, paresthesia, pruritus, rash, swelling, and urticaria.
Mild to moderate allergic reactions were observed in less than 1% of patients within approximately 30 min after plerixafor administration, including one or more of the following: urticaria (n=2), periorbital swelling (n=2), dyspnea (n=1) or hypoxia (n=1). Symptoms generally responded to treatments (e.g., antihistamines, corticosteroids, hydration or supplemental oxygen) or resolved spontaneously.
Vasovagal reactions, orthostatic hypotension, and/or syncope can occur following subcutaneous injections. In plerixafor oncology and healthy volunteer clinical studies, less than 1% of subjects experienced vasovagal reactions following subcutaneous administration of plerixafor doses ≤0.24 mg/kg. The majority of these events occurred within 1 hour of plerixafor administration. Because of the potential for these reactions, appropriate precautions should be taken.
Other adverse reactions in the randomized studies that occurred in <5% of patients but were reported as related to plerixafor during HSC mobilization and apheresis included abdominal pain, hyperhidrosis, abdominal distention, dry mouth, erythema, stomach discomfort, malaise, hypoesthesia oral, constipation, dyspepsia, and musculoskeletal pain.
Hyperleukocytosis: In clinical trials, white blood cell counts of 100,000/mcL or greater were observed, on the day prior to or any day of apheresis, in 7% of patients receiving plerixafor and in 1% of patients receiving placebo. No complications or clinical symptoms of leukostasis were observed.
6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been reported from postmarketing experience with plerixafor. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System: Splenomegaly and splenic rupture
Immune System Disorders: Anaphylactic reactions, including anaphylactic shock
Psychiatric Disorders: Abnormal dreams and nightmares -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited available data with plerixafor use in pregnant women are insufficient to inform a drug- associated risk of adverse developmental outcomes. In animal reproduction studies, subcutaneous administration of plerixafor to pregnant rats during organogenesis at doses 10-times the maximum recommended human doses resulted in embryo-fetal mortality, structural abnormalities, and alterations to growth [see Data].
Advise pregnant women of the potential risk to the fetus.
Advise women of reproductive potential to avoid becoming pregnant while receiving treatment with plerixafor [see Warnings and Precautions (5.6)].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriages for the indicated population is unknown.
Data
Animal data
Plerixafor administered to pregnant rats induced embryo-fetal toxicity, including fetal death, increased resorptions and postimplantation loss, decreased fetal weights, anophthalmia, shortened digits, cardiac interventricular septal defect, ringed aorta, globular heart, hydrocephaly, dilatation of olfactory ventricles, and retarded skeletal development. Embryo-fetal toxicities occurred mainly at a dose of 90 mg/m2 (approximately 10 times the recommended human dose of 0.24 mg/kg when compared on a mg/m2 basis).8.2 Lactation
Risk Summary
There are no data on the presence of plerixafor in human milk, the effect on the breastfed child, or the effect on milk production. Because of the potential serious adverse reactions in the breastfed child, advise females that breastfeeding is not recommended during treatment with plerixafor and for one week after the final dose.8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating plerixafor [see Use in Specific Populations (8.1)].
Contraception
Females
Plerixafor can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with plerixafor and for one week after the final dose.
Males
Males treated with plerixafor should use effective contraception during treatment and for one week after cessation of treatment.8.4 Pediatric Use
The safety and effectiveness of plerixafor have not been established in pediatric patients.
Effectiveness was not demonstrated in a single phase 1/2 randomized, open-label, comparative study of plerixafor plus standard regimens for mobilization of hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Forty-five pediatric patients ages 1 to <17 years with solid tumors or lymphoma were randomized 2:1 to plerixafor in addition to standard mobilization regimens (N=30) or standard mobilization regimens alone (N=15) (comparator arm). No new safety signals were observed in pediatric patients in this trial.
The day prior to the first apheresis, median peripheral blood CD34+ counts were 35 × 106 cells/L in the comparator arm and 15 × 106 cells/L in the plerixafor arm. On the day of the first apheresis, median peripheral blood CD34+ counts were 64 × 106 cells/L in the comparator arm and 77 × 106 cells/L in the plerixafor arm.
Plerixafor exposure, shown as AUC, in pediatric patients aged 12 to 18 years was within the range of values previously observed in adults, while the AUC of plerixafor in pediatric patients aged 2 to 12 years was 67% to 87% of that observed in adults, given the same adult dose (0.24 mg/kg).8.5 Geriatric Use
Of the total number of subjects in controlled clinical studies of plerixafor, 24% were 65 and over, while 0.8% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Since plerixafor is mainly excreted by the kidney, no dose modifications are necessary in elderly individuals with normal renal function. In general, care should be taken in dose selection for elderly patients due to the greater frequency of decreased renal function with advanced age. Dosage adjustment in elderly patients with CLCR less than or equal to 50 mL/min is recommended [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].8.6 Renal Impairment
In patients with moderate and severe renal impairment (CLCR less than or equal to 50 mL/min), reduce the dose of plerixafor by one-third to 0.16 mg/kg [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Plerixafor injection is a sterile, preservative-free, clear, colorless to pale-yellow, isotonic solution for subcutaneous injection. Each mL of the sterile solution contains 20 mg of plerixafor. Each single-dose vial is filled to deliver 1.2 mL of the sterile solution that contains 24 mg of plerixafor and 5.9 mg of sodium chloride in Water for Injection adjusted to a pH of 6.0 to 7.5 with hydrochloric acid and with sodium hydroxide, if required.
Plerixafor is a hematopoietic stem cell mobilizer with a chemical name 1,4-Bis((1,4,8,11-tetraazacyclotetradecan-1-yl)methyl)benzene. It has the molecular formula C28H54N8. The molecular weight of plerixafor is 502.79 g/mol. The structural formula is provided in Figure 1.
Figure 1: Structural Formula
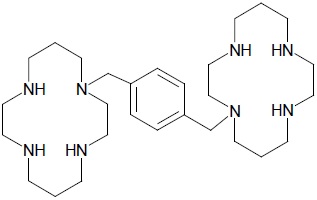
Plerixafor is a white to off-white powder. It is hygroscopic. Plerixafor has a typical melting point of 131.5°C. The partition coefficient of plerixafor between 1-octanol and pH 7 aqueous buffer is <0.1. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Plerixafor is an inhibitor of the CXCR4 chemokine receptor and blocks binding of its cognate ligand, stromal cell-derived factor-1α (SDF-1α). SDF-1α and CXCR4 are recognized to play a role in the trafficking and homing of human hematopoietic stem cells (HSCs) to the marrow compartment. Once in the marrow, stem cell CXCR4 can act to help anchor these cells to the marrow matrix, either directly via SDF-1α or through the induction of other adhesion molecules. Treatment with plerixafor resulted in leukocytosis and elevations in circulating hematopoietic progenitor cells in mice, dogs and humans. CD34+ cells mobilized by plerixafor were capable of engraftment with long-term repopulating capacity up to one year in canine transplantation models.
12.2 Pharmacodynamics
Data on the fold increase in peripheral blood CD34+ cell count (cells/mcL) by apheresis day were evaluated in two placebo-controlled clinical studies in patients with NHL and MM (Study 1 and Study 2, respectively). The fold increase in CD34+ cell count (cells/mcL) over the 24-hour period starting from the day prior to the first apheresis and ending the next morning just before the first apheresis is summarized in Table 3. During this 24-hour period, a single dose of plerixafor or placebo was administered 10 to 11 hours prior to apheresis.
Table 3: Fold Increase in Peripheral Blood CD34+ Cell Count Following Pretreatment with Filgrastim and Administration of Plerixafor Study
Plerixafor and Filgrastim
Placebo and Filgrastim
Median
Mean (SD)
Median
Mean (SD)
Study 1
5
6.1 (5.4)
1.4
1.9 (1.5)
Study 2
4.8
6.4 (6.8)
1.7
2.4 (7.3)
In pharmacodynamic studies of plerixafor in healthy volunteers, peak mobilization of CD34+ cells was observed between 6 and 9 hours after administration. In pharmacodynamic studies of plerixafor in conjunction with filgrastim in healthy volunteers, a sustained elevation in the peripheral blood CD34+ count was observed from 4 to 18 hours after plerixafor administration with a peak CD34+ count between 10 and 14 hours.
QT/QTc Prolongation
There is no indication of a QT/QTc prolonging effect of plerixafor in single doses up to 0.40 mg/kg. In a randomized, double-blind, crossover study, 48 healthy subjects were administered a single subcutaneous dose of plerixafor (0.24 mg/kg and 0.40 mg/kg) and placebo. Peak concentrations for 0.40 mg/kg plerixafor were approximately 1.8-fold higher than the peak concentrations following the 0.24 mg/kg single subcutaneous dose.12.3 Pharmacokinetics
The single-dose pharmacokinetics of plerixafor 0.24 mg/kg were evaluated in patients with NHL and MM following pretreatment with filgrastim (10 mcg/kg once daily for 4 consecutive days). Plerixafor exhibits linear kinetics between the 0.04 mg/kg to 0.24 mg/kg dose range. The pharmacokinetics of plerixafor was similar across clinical studies in healthy subjects who received plerixafor alone and NHL and MM patients who received plerixafor in combination with filgrastim.
A population pharmacokinetic analysis incorporated plerixafor data from 63 subjects (NHL patients, MM patients, subjects with varying degrees of renal impairment, and healthy subjects) who received a single subcutaneous dose (0.04 mg/kg to 0.24 mg/kg) of plerixafor. A two-compartment disposition model with first order absorption and elimination was found to adequately describe the plerixafor concentration-time profile. Significant relationships between clearance and creatinine clearance (CLCR), as well as between central volume of distribution and body weight were observed. The distribution half-life (t1/2α) was estimated to be 0.3 hours and the terminal population half-life (t1/2β) was 5.3 hours in patients with normal renal function.
The population pharmacokinetic analysis showed that the mg/kg-based dosage results in an increased plerixafor exposure (AUC0-24h) with increasing body weight. In order to compare the pharmacokinetics and pharmacodynamics of plerixafor following 0.24 mg/kg-based and fixed (20 mg) doses, a follow-up trial was conducted in patients with NHL (N=61) who were treated with 0.24 mg/kg or 20 mg of plerixafor. The trial was conducted in patients weighing 70 kg or less. The fixed 20 mg dose showed 1.43-fold higher exposure (AUC0-10h) than the 0.24 mg/kg dose (Table 4). The fixed 20 mg dose also showed numerically higher response rate (5.2% [60% vs 54.8%] based on the local lab data and 11.7% [63.3% vs 51.6%] based on the central lab data) in attaining the target of ≥5 × 106 CD34+ cells/kg than the mg/kg-based dose. However, the median time to reach ≥5 × 106 CD34+ cells/kg was 3 days for both treatment groups, and the safety profile between the groups was similar. Based on these results, further analysis was conducted by FDA reviewers and a body weight of 83 kg was selected as an appropriate cut-off point to transition patients from fixed to weight based dosing.
Table 4: Systemic Exposure (AUC0-10h) Comparisons of Fixed and Weight-Based Regimens Regimen
Geometric Mean AUC
Fixed 20 mg (n=30)
3991.2
0.24 mg/kg (n=31)
2792.7
Ratio (90% CI)
1.43 (1.32,1.54)
There is limited experience with the 0.24 mg/kg dose of plerixafor in patients weighing above 160 kg. Therefore, the dose should not exceed that of a 160 kg patient (i.e., 40 mg/day if CLCR is greater than 50 mL/min and 27 mg/day if CLCR is less than or equal to 50 mL/min) [see Dosage and Administration (2.1, 2.3)].
Absorption
Peak plasma concentrations occurred at approximately 30 to 60 minutes after a subcutaneous dose.
Distribution
Plerixafor is bound to human plasma proteins up to 58%. The apparent volume of distribution of plerixafor in humans is 0.3 L/kg demonstrating that plerixafor is largely confined to, but not limited to, the extravascular fluid space.
Metabolism
The metabolism of plerixafor was evaluated with in vitro assays. Plerixafor is not metabolized as shown in assays using human liver microsomes or human primary hepatocytes and does not exhibit inhibitory activity in vitro towards the major drug metabolizing cytochrome P450 enzymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4/5). In in vitro studies with human hepatocytes, plerixafor does not induce CYP1A2, CYP2B6, or CYP3A4 enzymes. These findings suggest that plerixafor has a low potential for involvement in cytochrome P450-dependent drug-drug interactions.
Elimination
The major route of elimination of plerixafor is urinary. Following a 0.24 mg/kg dose in healthy volunteers with normal renal function, approximately 70% of the dose was excreted in the urine as the parent drug during the first 24 hours following administration. In studies with healthy subjects and patients, the terminal half-life in plasma ranges between 3 and 5 hours. At concentrations similar to what are seen clinically, plerixafor did not act as a substrate or inhibitor of P-glycoprotein in an in vitro study with MDCKII and MDCKII-MDR1 cell models.
Special Populations
Renal impairment
Following a single 0.24 mg/kg subcutaneous dose, plerixafor clearance was reduced in subjects with varying degrees of renal impairment and was positively correlated with CLCR. The mean AUC0 24h of plerixafor in subjects with mild (CLCR 51 to 80 mL/min), moderate (CLCR 31 to 50 mL/min), and severe (CLCR <31 mL/min) renal impairment was 7%, 32%, and 39% higher than healthy subjects with normal renal function, respectively. Renal impairment had no effect on Cmax. A population pharmacokinetic analysis indicated an increased exposure (AUC0-24h) in patients with moderate and severe renal impairment compared to patients with CLCR >50 mL/min. These results support a dose reduction of one-third in patients with moderate to severe renal impairment (CLCR ≤50 mL/min) in order to match the exposure in patients with normal renal function. The population pharmacokinetic analysis showed that the mg/kg-based dosage results in an increased plerixafor exposure (AUC0-24h) with increasing body weight; therefore, if CLCR is ≤50 mL/min the dose should not exceed 27 mg/day [see Dosage and Administration (2.3)].
Since plerixafor is primarily eliminated by the kidneys, coadministration of plerixafor with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of plerixafor or the coadministered drug. The effects of coadministration of plerixafor with other drugs that are renally eliminated or are known to affect renal function have not been evaluated.
Race
Clinical data show similar plerixafor pharmacokinetics for Caucasians and African Americans, and the effect of other racial/ethnic groups has not been studied.
Gender
Clinical data show no effect of gender on plerixafor pharmacokinetics.
Age
Clinical data show no effect of age on plerixafor pharmacokinetics.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with plerixafor have not been conducted.
Plerixafor was not genotoxic in an in vitro bacterial mutation assay (Ames test in Salmonella), an in vitro chromosomal aberration test using V79 Chinese hamster cells, or an in vivo bone marrow micronucleus test in rats after subcutaneous doses up to 25 mg/kg (150 mg/m2).
The effect of plerixafor on human fertility is unknown. The effect of plerixafor on male fertility was not studied in designated reproductive toxicology studies. The staging of spermatogenesis measured in a 28-day repeated dose toxicity study in rats revealed no abnormalities considered to be related to plerixafor. No histopathological evidence of toxicity to male or female reproductive organs was observed in 28-day repeated dose toxicity studies.
No adverse effects on estrus or reproductive indices were observed in an investigative fertility study in female rats administered plerixafor at doses up to 90 mg/m2 (15 mg/kg/day) or approximately 10 times the recommended human dose of 0.24 mg/kg when compared on a mg/m2 basis. -
14 CLINICAL STUDIES
The efficacy and safety of plerixafor in conjunction with filgrastim in non-Hodgkin’s lymphoma (NHL) Study AMD 3100-3101 (referred to as study 1) (NCT00103610) and multiple myeloma (MM) Study AMD 3100-3102 (referred to as study 2) (NCT00103662) were evaluated in two placebo-controlled studies (Studies 1 and 2). Patients were randomized to receive either plerixafor 0.24 mg/kg or placebo on each evening prior to apheresis. Patients received daily morning doses of filgrastim 10 mcg/kg for 4 days prior to the first dose of plerixafor or placebo and on each morning prior to apheresis. Two hundred and ninety-eight (298) NHL patients were included in the primary efficacy analyses for Study 1. The mean age was 55 years (range 29 to 75) and 58 years (range 22 to 75) in the plerixafor and placebo groups, respectively, and 93% of subjects were Caucasian. In study 2, 302 patients with MM were included in the primary efficacy analyses. The mean age (58 years) and age range (28 to 75) were similar in the plerixafor and placebo groups, and 81% of subjects were Caucasian.
In Study 1, 59% of NHL patients who were mobilized with plerixafor and filgrastim collected ≥5 × 106 CD34+ cells/kg from the peripheral blood in four or fewer apheresis sessions, compared with 20% of patients who were mobilized with placebo and filgrastim (p <0.001). Other CD34+ cell mobilization outcomes showed similar findings (Table 5).
Table 5: Study 1 Efficacy Results - CD34+ Cell Mobilization in NHL Patients * p-value calculated using Pearson’s Chi-Squared test Efficacy Endpoint
Plerixafor
and Filgrastim
(n=150)
Placebo and
Filgrastim
(n=148)
p-value*
Patients achieving ≥5 × 106 cells/kg in ≤4 apheresis days
89 (59%)
29 (20%)
<0.001
Patients achieving ≥2 × 106 cells/kg in ≤4 apheresis days
130 (87%)
70 (47%)
<0.001
The median number of days to reach ≥5 × 106 CD34+ cells/kg was 3 days for the plerixafor group and not evaluable for the placebo group. Table 6 presents the proportion of patients who achieved ≥5 × 106 CD34+ cells/kg by apheresis day.
Table 6: Study 1 Efficacy Results – Proportion of Patients Who Achieved ≥5 × 106 CD34+ cells/kg by Apheresis Day in NHL Patients * Percents determined by Kaplan Meier method
† n includes all patients who received at least one day of apheresisDays
Proportion*
in Plerixafor and Filgrastim
(n=147†)
Proportion*
in Placebo and Filgrastim
(n=142†)
1
27.9%
4.2%
2
49.1%
14.2%
3
57.7%
21.6%
4
65.6%
24.2%
In Study 2, 72% of MM patients who were mobilized with plerixafor and filgrastim collected ≥6 × 106 CD34+ cells/kg from the peripheral blood in two or fewer apheresis sessions, compared with 34% of patients who were mobilized with placebo and filgrastim (p <0.001). Other CD34+ cell mobilization outcomes showed similar findings (Table 7).Table 7: Study 2 Efficacy Results – CD34+ Cell Mobilization in Multiple Myeloma Patients * p-value calculated using Pearson’s Chi-Squared test Efficacy Endpoint
Plerixafor and
Filgrastim
(n=148)
Placebo and
Filgrastim
(n=154)
p-value*
Patients achieving ≥6 × 106 cells/kg in ≤2 apheresis days
106 (72%)
53 (34%)
<0.001
Patients achieving ≥6 × 106 cells/kg in ≤4 apheresis days
112 (76%)
79 (51%)
<0.001
Patients achieving ≥2 × 106 cells/kg in ≤4 apheresis days
141 (95%)
136 (88%)
0.028
The median number of days to reach ≥6 × 106 CD34+ cells/kg was 1 day for the plerixafor group and 4 days for the placebo group. Table 8 presents the proportion of patients who achieved ≥6 × 106 CD34+ cells/kg by apheresis day.
Table 8: Study 2 – Proportion of Patients Who Achieved ≥6 × 106 CD34+ cells/kg by Apheresis Day in MM Patients * Percents determined by Kaplan Meier method
† n includes all patients who received at least one day of apheresisDays
Proportion*
in Plerixafor and Filgrastim
(n=144†)
Proportion*
in Placebo and Filgrastim
(n=150†)
1
54.2%
17.3%
2
77.9%
35.3%
3
86.8%
48.9%
4
86.8%
55.9%
Multiple factors can influence time to engraftment and graft durability following stem cell transplantation. For transplanted patients in the Phase 3 studies, time to neutrophil and platelet engraftment and graft durability were similar across the treatment groups.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Plerixafor injection, 24 mg/1.2 mL (20 mg/mL) is a sterile, preservative-free, clear, colorless to pale-yellow solution supplied in a 4 mL clear glass single-dose vial.
24 mg per 1.2 mL (20 mg/mL)
Single-Dose Vial
Packaged Individually NDC 55150-356-01
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
The vial stopper is not made with natural rubber latex. -
17 PATIENT COUNSELING INFORMATION
Advise patients of the potential for anaphylactic reactions, including signs and symptoms such as urticaria, periorbital swelling, dyspnea, or hypoxia during and following plerixafor injection and to report these symptoms immediately to a healthcare professional [see Adverse Reactions (6.1, 6.2)].
Advise patients to contact healthcare professional immediately if they experience left upper abdominal pain and/or scapular or shoulder pain [see Adverse Reactions (6.1, 6.2)].
Advise patients to inform a healthcare professional immediately if symptoms of vasovagal reactions such as orthostatic hypotension or syncope occur during or shortly after their plerixafor injection [see Adverse Reactions (6.1)].
Advise patients who experience itching, rash, or reaction at the site of injection to notify a healthcare professional, as these symptoms have been treated with over-the-counter medications during clinical trials [see Adverse Reactions (6.1)].
Advise patients that plerixafor may cause gastrointestinal disorders, including diarrhea, nausea, vomiting, flatulence, and abdominal pain. Patients should be told how to manage specific gastrointestinal disorders and to inform their healthcare professional if severe events occur following plerixafor injection [see Adverse Reactions (6.1)].
Advise females of reproductive potential of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with plerixafor [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Advise females and males of reproductive potential to use effective contraceptive methods during plerixafor use and for 1 week following cessation of treatment[see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Advise women not to breastfeed during treatment with plerixafor and for 1 week following the last dose [Use in Specific Populations (8.2)].
Distributed by:
Eugia US LLC
279 Princeton-Hightstown Rd.
E. Windsor, NJ 08520
Manufactured by:
Eugia Pharma Specialities Limited
Hyderabad - 500032
India - PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 24 mg per 1.2 mL (20 mg/mL) Container Label
- PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 24 mg per 1.2 mL (20 mg/mL) Container-Carton Label
-
INGREDIENTS AND APPEARANCE
PLERIXAFOR
plerixafor solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55150-356 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PLERIXAFOR (UNII: S915P5499N) (PLERIXAFOR - UNII:S915P5499N) PLERIXAFOR 24 mg in 1.2 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55150-356-01 1 in 1 CARTON 07/24/2023 1 1.2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213672 07/24/2023 Labeler - Eugia US LLC (968961354) Establishment Name Address ID/FEI Business Operations EUGIA Pharma Specialities Limited 872201704 ANALYSIS(55150-356) , MANUFACTURE(55150-356) , PACK(55150-356)