Label: SPINRAZA- nusinersen injection, solution


- NDC Code(s): 64406-058-01
- Packager: Biogen
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated April 24, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use
SPINRAZA® safely and effectively. See full prescribing information for SPINRAZA.
SPINRAZA (nusinersen) injection, for intrathecal use
Initial U.S. Approval: 2016INDICATIONS AND USAGE
SPINRAZA is a survival motor neuron-2 (SMN2)-directed antisense oligonucleotide indicated for the treatment of spinal muscular atrophy (SMA) in pediatric and adult patients (1)
DOSAGE AND ADMINISTRATION
SPINRAZA is administered intrathecally (2.1)
Dosing Information (2.1)
- The recommended dosage is 12 mg (5 mL) per administration
- Initiate SPINRAZA treatment with 4 loading doses: the first three loading doses should be administered at 14-day intervals; the 4th loading dose should be administered 30 days after the 3rd dose. A maintenance dose should be administered once every 4 months thereafter.
Important Preparation and Administration Instructions (2.2)
- Allow to warm to room temperature prior to administration
- Administer within 4 hours of removal from vial
- Prior to administration, remove 5 mL of cerebrospinal fluid
- Administer as intrathecal bolus injection over 1 to 3 minutes
Laboratory Testing and Monitoring to Assess Safety (2.3)
- At baseline and prior to each dose, obtain a platelet count, coagulation laboratory testing, and quantitative spot urine protein testing
DOSAGE FORMS AND STRENGTHS
Injection: 12 mg/5 mL (2.4 mg/mL) in a single-dose vial (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most common adverse reactions that occurred in at least 20% of SPINRAZA-treated patients and occurred at least 5% more frequently than in control patients were:
- lower respiratory infection and constipation in patients with infantile-onset SMA (6.1)
- pyrexia, headache, vomiting, and back pain in patients with later-onset SMA (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-844-477-4672 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Important Preparation and Administration Instructions
2.3 Laboratory Testing and Monitoring to Assess Safety
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombocytopenia and Coagulation Abnormalities
5.2 Renal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Infantile-Onset SMA
14.2 Later-Onset SMA
14.3 Presymptomatic SMA
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
SPINRAZA is administered intrathecally by, or under the direction of, healthcare professionals experienced in performing lumbar punctures.
Recommended Dosage
The recommended dosage is 12 mg (5 mL) per administration.
Initiate SPINRAZA treatment with 4 loading doses. The first three loading doses should be administered at 14-day intervals. The 4th loading dose should be administered 30 days after the 3rd dose. A maintenance dose should be administered once every 4 months thereafter.
Missed Dose
Missed Loading Dose
If a loading dose (any of the 4 loading doses) is missed, administer the missed loading dose as soon as possible; adjust the date for the subsequent doses to maintain the recommended interval between doses.
Missed Maintenance Dose
Less than 8 months from last dose
Administer the missed maintenance dose as soon as possible; then administer the next maintenance dose per the originally scheduled date, as long as these two doses are administered at least 14 days apart.
At least 8 months but less than 16 months from last dose
Administer the missed maintenance dose as soon as possible, followed by one additional dose 14 days later, and then administer the next maintenance dose 4 months thereafter.
2.2 Important Preparation and Administration Instructions
SPINRAZA is for intrathecal use only.
Prepare and use SPINRAZA according to the following steps using aseptic technique. Each vial is intended for single dose only.
Preparation
- Store SPINRAZA in the carton in a refrigerator until time of use.
- Allow the SPINRAZA vial to warm to room temperature (25o C/77o F) prior to administration. Do not use external heat sources.
- Inspect the SPINRAZA vial for particulate matter and discoloration prior to administration. Do not administer SPINRAZA if visible particulates are observed or if the liquid in the vial is discolored. The use of external filters is not required.
- Withdraw 12 mg (5 mL) of SPINRAZA from the single-dose vial into a syringe and discard unused contents of the vial.
- Administer SPINRAZA within 4 hours of removal from vial.
Administration
- Consider sedation as indicated by the clinical condition of the patient.
- Consider ultrasound or other imaging techniques to guide intrathecal administration of SPINRAZA, particularly in younger patients.
- Prior to administration, remove 5 mL of cerebrospinal fluid.
- Administer SPINRAZA as an intrathecal bolus injection over 1 to 3 minutes using a spinal anesthesia needle [see Dosage and Administration (2.1)]. Do not administer SPINRAZA in areas of the skin where there are signs of infection or inflammation [see Adverse Reactions (6.3)].
2.3 Laboratory Testing and Monitoring to Assess Safety
Conduct the following laboratory tests at baseline and prior to each dose of SPINRAZA and as clinically needed [see Warnings and Precautions (5.1, 5.2)]:
- Platelet count
- Prothrombin time; activated partial thromboplastin time
- Quantitative spot urine protein testing
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombocytopenia and Coagulation Abnormalities
Coagulation abnormalities and thrombocytopenia, including acute severe thrombocytopenia, have been observed after administration of some antisense oligonucleotides.
In the sham-controlled studies for patients with infantile-onset and later-onset SMA, 24 of 146 (16%) SPINRAZA-treated patients with high, normal, or unknown platelet count at baseline developed a platelet level below the lower limit of normal, compared to 10 of 72 (14%) sham-controlled patients.
In the sham-controlled study in patients with later-onset SMA (Study 2), two SPINRAZA-treated patients developed platelet counts less than 50,000 cells per microliter, with a lowest level of 10,000 cells per microliter recorded on study day 28.
Because of the risk of thrombocytopenia and coagulation abnormalities from SPINRAZA, patients may be at increased risk of bleeding complications.
Perform a platelet count and coagulation laboratory testing at baseline and prior to each administration of SPINRAZA and as clinically needed.
5.2 Renal Toxicity
Renal toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.
SPINRAZA is present in and excreted by the kidney [see Clinical Pharmacology (12.3)]. In the sham-controlled studies for patients with infantile-onset and later-onset SMA, 71 of 123 (58%) of SPINRAZA-treated patients had elevated urine protein, compared to 22 of 65 (34%) sham-controlled patients. Conduct quantitative spot urine protein testing (preferably using a first morning urine specimen) at baseline and prior to each dose of SPINRAZA. For urinary protein concentration greater than 0.2 g/L, consider repeat testing and further evaluation.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described in detail in other sections of the labeling:
- Thrombocytopenia and Coagulation Abnormalities [see Warnings and Precautions (5.1)]
- Renal Toxicity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of SPINRAZA cannot be directly compared to rates in clinical trials of other drugs and may not reflect the rates observed in practice.
In clinical studies, 346 patients (47% male, 76% Caucasian) were treated with SPINRAZA, including 314 exposed for at least 6 months, 258 exposed for at least 1 year, and 138 exposed for at least 2 years. The safety of SPINRAZA was studied in presymptomatic infants with SMA; pediatric patients (approximately 3 days to 16 years of age at first dose) with symptomatic SMA; in a sham-controlled trial in infants with symptomatic SMA (Study 1; n=80 for SPINRAZA, n=41 for control); in a sham-controlled trial in children with symptomatic SMA (Study 2; n=84 for SPINRAZA, n=42 for control); in an open-label study in presymptomatic infants (Study 3, n=25) and other studies in symptomatic infants (n=54) and later-onset patients (n=103). In Study 1, 58 patients were exposed for at least 6 months and 28 patients were exposed for at least 12 months. In Study 2, 84 patients were exposed for at least 6 months and 82 patients were exposed for at least 12 months.
Clinical Trial in Infantile-Onset SMA (Study 1)
In Study 1, baseline disease characteristics were largely similar in the SPINRAZA-treated patients and sham-control patients except that SPINRAZA-treated patients at baseline had a higher percentage compared to sham-control patients of paradoxical breathing (89% vs 66%), pneumonia or respiratory symptoms (35% vs 22%), swallowing or feeding difficulties (51% vs 29%), and requirement for respiratory support (26% vs 15%).
The most common adverse reactions that occurred in at least 20% of SPINRAZA-treated patients and occurred at least 5% more frequently than in control patients were lower respiratory infection and constipation. Serious adverse reactions of atelectasis were more frequent in SPINRAZA-treated patients (18%) than in control patients (10%). Because patients in Study 1 were infants, adverse reactions that are verbally reported could not be assessed in this study.
Table 1. Adverse Reactions that Occurred in at Least 5% of SPINRAZA Patients and Occurred at Least 5% More Frequently or At Least 2 Times as Frequently Than in Control Patients with Infantile-Onset SMA (Study 1) 1 Loading doses followed by 12 mg (5 mL) once every 4 months
2 Includes adenovirus infection, bronchiolitis, bronchitis, bronchitis viral, corona virus infection, Influenza, lower respiratory tract infection, lower respiratory tract infection viral, lung infection, parainfluenzae virus infection, pneumonia, pneumonia bacterial, pneumonia influenzal, pneumonia moraxella, pneumonia parainfluenzae viral, pneumonia pneumococcal, pneumonia pseudomonal, pneumonia respiratory syncytial viral, pneumonia viral, and respiratory syncytial virus bronchiolitis.
Adverse Reactions SPINRAZA 12 mg1
N = 80
%Sham-Procedure Control
N = 41
%Lower respiratory infection2 55 37 Constipation 35 22 Teething 18 7 Urinary tract infection 9 0 Upper respiratory tract congestion 8 2 Ear infection 6 2 Flatulence 5 2 Decreased weight 5 2 In an open-label clinical study in infants with symptomatic SMA, severe hyponatremia was reported in a patient treated with SPINRAZA requiring salt supplementation for 14 months.
Cases of rash were reported in patients treated with SPINRAZA. One patient, 8 months after starting SPINRAZA treatment, developed painless red macular lesions on the forearm, leg, and foot over an 8-week period. The lesions ulcerated and scabbed over within 4 weeks, and resolved over several months. A second patient developed red macular skin lesions on the cheek and hand ten months after the start of SPINRAZA treatment, which resolved over 3 months. Both cases continued to receive SPINRAZA and had spontaneous resolution of the rash.
SPINRAZA may cause a reduction in growth as measured by height when administered to infants, as suggested by observations from the controlled study. It is unknown whether any effect of SPINRAZA on growth would be reversible with cessation of treatment.
Clinical Trial in Later-Onset SMA (Study 2)
In Study 2, baseline disease characteristics were largely similar in the SPINRAZA-treated patients and sham-control patients except for the proportion of SPINRAZA-treated patients who had ever achieved the ability to stand without support (13% vs 29%) or walk with support (24% vs 33%).
The most common adverse reactions that occurred in at least 20% of SPINRAZA-treated patients and occurred at least 5% more frequently than in control patients were pyrexia, headache, vomiting, and back pain.
Table 2. Adverse Reactions that Occurred in at Least 5% of SPINRAZA Patients and Occurred at Least 5% More Frequently or At Least 2 Times as Frequently Than in Control Patients with Later-Onset SMA (Study 2) 1 Loading doses followed by 12 mg (5 mL) once every 6 months
Adverse Reactions SPINRAZA 12 mg1
N=84
%Sham-Procedure Control
N=42
%Pyrexia 43 36 Headache 29 7 Vomiting 29 12 Back pain 25 0 Epistaxis 7 0 Fall 5 0 Respiratory tract congestion 5 2 Seasonal allergy 5 2 Post-lumbar puncture syndrome has also been observed after administration of SPINRAZA.
6.2 Immunogenicity
As with all oligonucleotides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to nusinersen in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The immunogenic response to nusinersen was evaluated in 294 patients with post-baseline plasma samples for anti-drug antibodies (ADAs). Seventeen patients (6%) developed treatment-emergent ADAs, of which 5 were transient, 12 were considered to be persistent. Persistent was defined as having one positive test followed by another one more than 100 days after the first positive test. In addition, “persistent” is also defined as having one or more positive samples and no sample more than 100 days after the first positive sample. Transient was defined as having one or more positive results and not confirmed to be persistent. There are insufficient data to evaluate an effect of ADAs on clinical response, adverse events, or the pharmacokinetic profile of nusinersen.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of SPINRAZA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Serious infections associated with lumbar puncture, such as meningitis, have been reported. Hydrocephalus, aseptic meningitis, hypersensitivity reactions (e.g. angioedema, urticaria, rash), and arachnoiditis have also been reported.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of SPINRAZA in pregnant women. When nusinersen was administered by subcutaneous injection to mice throughout pregnancy and lactation, developmental toxicity (long-term neurobehavioral impairment) was observed at all doses tested (see Data). In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
When nusinersen (0, 3, 10, or 25 mg/kg) was administered subcutaneously to male and female mice every other day prior to and during mating and continuing in females throughout organogenesis, no adverse effects on embryofetal development were observed. Subcutaneous administration of nusinersen (0, 6, 12.6, or 25 mg/kg) to pregnant rabbits every other day throughout organogenesis produced no evidence of embryofetal developmental toxicity.
When nusinersen (1.4, 5.8, or 17.2 mg/kg) was administered to pregnant female mice by subcutaneous injection every other day throughout organogenesis and continuing once every six days throughout the lactation period, adverse neurobehavioral effects (alterations in locomotor activity, learning and memory deficits) were observed when offspring were tested after weaning or as adults. A no-effect level for neurobehavioral impairment was not established.
8.2 Lactation
Risk Summary
There are no data on the presence of nusinersen in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.Nusinersen was detected in the milk of lactating mice when administered by subcutaneous injection. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SPINRAZA and any potential adverse effects on the breastfed infant from SPINRAZA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SPINRAZA in pediatric patients from newborn to 17 years have been established [see Clinical Studies (14.1)].
Juvenile Animal Toxicity Data
In intrathecal toxicity studies in juvenile monkeys, administration of nusinersen (0, 0.3, 1, or 3 mg/dose for 14 weeks and 0, 0.3, 1, or 4 mg/dose for 53 weeks) resulted in brain histopathology (neuronal vacuolation and necrosis/cellular debris in the hippocampus) at the mid and high doses and acute, transient deficits in lower spinal reflexes at the high dose in each study. In addition, possible neurobehavioral deficits were observed on a learning and memory test at the high dose in the 53-week monkey study. The no-effect dose for neurohistopathology in monkeys (0.3 mg/dose) is approximately equivalent to the human dose when calculated on a yearly basis and corrected for the species difference in CSF volume.
-
11 DESCRIPTION
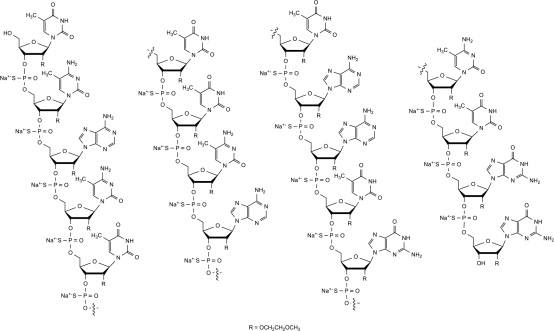
Nusinersen is a modified antisense oligonucleotide, where the 2'-hydroxy groups of the ribofuranosyl rings are replaced with 2'-O-2-methoxyethyl groups and the phosphate linkages are replaced with phosphorothioate linkages. Nusinersen binds to a specific sequence in the intron downstream of exon 7 of the SMN2 transcript. The structural formula is:
SPINRAZA is supplied as a sterile, preservative-free, colorless solution for intrathecal use in a single-dose glass vial. Each 1 mL solution contains 2.4 mg of nusinersen (equivalent to 2.53 mg of nusinersen sodium salt). Each 1 mL also contains calcium chloride dihydrate (0.21 mg) USP, magnesium chloride hexahydrate (0.16 mg) USP, potassium chloride (0.22 mg) USP, sodium chloride (8.77 mg) USP, sodium phosphate dibasic anhydrous (0.10 mg) USP, sodium phosphate monobasic dihydrate (0.05 mg) USP, and Water for Injection USP. The product may contain hydrochloric acid or sodium hydroxide to adjust pH. The pH is ~7.2.
The molecular formula of SPINRAZA is C234H323N61O128P17S17Na17 and the molecular weight is 7501.0 daltons.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
SPINRAZA is an antisense oligonucleotide (ASO) designed to treat SMA caused by mutations in chromosome 5q that lead to SMN protein deficiency. Using in vitro assays and studies in transgenic animal models of SMA, SPINRAZA was shown to increase exon 7 inclusion in SMN2 messenger ribonucleic acid (mRNA) transcripts and production of full-length SMN protein.
12.2 Pharmacodynamics
Autopsy samples from patients (n=3) had higher levels of SMN2 messenger ribonucleic acid (mRNA) containing exon 7 in the thoracic spinal cord compared to untreated SMA infants.
Cardiac Electrophysiology
Across the sham-controlled studies in 247 patients with spinal muscular atrophy who received either SPINRAZA or sham-control, QTcF values >500 ms and change from baseline values >60 ms were observed in 4 (2.4%) patients receiving SPINRAZA. Compared to the sham-control, there was no increase in the incidence of cardiac adverse reactions associated with delayed ventricular repolarization in patients treated with SPINRAZA.
12.3 Pharmacokinetics
Absorption
Intrathecal injection of SPINRAZA into the cerebrospinal fluid (CSF) allows nusinersen to be distributed from the CSF to the target central nervous system (CNS) tissues. Following intrathecal administration, trough plasma concentrations of nusinersen were relatively low, compared to the trough CSF concentration. Median plasma Tmax values ranged from 1.7 to 6.0 hours. Mean plasma Cmax and AUC values increased approximately dose-proportionally up to a dose of 12 mg.
Distribution
Autopsy data from patients (n=3) showed that SPINRAZA administered intrathecally was distributed within the CNS and peripheral tissues, such as skeletal muscle, liver, and kidney.
Metabolism
Nusinersen is metabolized via exonuclease (3'- and 5')-mediated hydrolysis and is not a substrate for, or inhibitor or inducer of CYP450 enzymes.
Excretion
The mean terminal elimination half-life is estimated to be 135 to 177 days in CSF, and 63 to 87 days in plasma. The primary route of elimination is likely by urinary excretion for nusinersen and its chain-shortened metabolites. At 24 hours, only 0.5% of the administered dose was recovered in the urine.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Administration of nusinersen (0, 5, 15, or 50 mg/kg) to male and female mice by subcutaneous injection, once every two weeks for 2 years, resulted in an increase in the incidence of vascular tumors (combined hemangioma and hemangiosarcoma) at the highest dose tested.
-
14 CLINICAL STUDIES
The efficacy of SPINRAZA was demonstrated in two double-blind, sham-procedure controlled clinical trials in symptomatic infantile-onset and later-onset SMA patients (Study 1 and Study 2) and was supported by open-label clinical trials conducted in presymptomatic and symptomatic SMA patients. The overall findings from these trials support the effectiveness of SPINRAZA across the range of SMA patients, and appear to support the early initiation of treatment with SPINRAZA.
14.1 Infantile-Onset SMA
Study 1 (NCT02193074) was a multicenter, randomized, double-blind, sham-procedure controlled study in 121 symptomatic infants ≤ 7 months of age at the time of first dose, diagnosed with SMA (symptom onset before 6 months of age). Patients were randomized 2:1 to receive either 12 mg SPINRAZA or sham injection as a series of loading doses administered intrathecally followed by maintenance doses administered every 4 months. Patients in this study were deemed most likely to develop Type 1 SMA.
A planned interim efficacy analysis was conducted based on patients who died, withdrew, or completed at least 183 days of treatment. Of the 82 patients included in the interim analysis (52 patients in the SPINRAZA-treated group and 30 in the sham-control group), 44% were male, 87% were Caucasian, 2% were Black, and 4% were Asian. Age at first treatment ranged from 30 to 262 days (median 181). Length of treatment ranged from 6 to 442 days (median 261 days). Baseline demographics were balanced between the SPINRAZA and control groups with the exception of age at first treatment (median age 175 vs. 206 days, respectively). The SPINRAZA and control groups were balanced with respect to gestational age, birth weight, disease duration, and SMN2 copy number. Median disease duration was 14 weeks. There was some imbalance in age at symptom onset with 88% of subjects in the SPINRAZA group and 77% in the control group experiencing symptoms within the first 12 weeks of life.
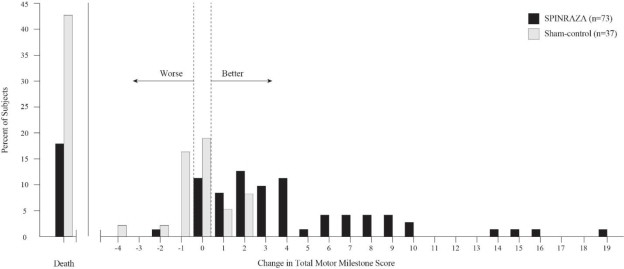
The primary endpoint assessed at the time of interim analysis was the proportion of responders: patients with an improvement in motor milestones according to Section 2 of the Hammersmith Infant Neurologic Exam (HINE). This endpoint evaluates seven different areas of motor milestone development, with a maximum score between 2-4 points for each, depending on the milestone, and a total maximum score of 26. A treatment responder was defined as any patient with at least a 2-point increase (or maximal score of 4) in ability to kick (consistent with improvement by at least 2 milestones), or at least a 1-point increase in the motor milestones of head control, rolling, sitting, crawling, standing or walking (consistent with improvement by at least 1 milestone). To be classified as a responder, patients needed to exhibit improvement in more categories of motor milestones than worsening. Of the 82 patients who were eligible for the interim analysis, a statistically significantly greater percentage of patients achieved the definition of a motor milestone responder in the SPINRAZA group (40%) compared to the sham-control group (0%). Results from the final analysis were consistent with those from the interim analysis (Table 3). Fifty-one percent of patients in the SPINRAZA group achieved the definition of a motor milestone responder compared to 0% of patients in the sham-control group. Figure 1 is a descriptive display of the distribution of net change from baseline in the total motor milestone score for Section 2 of the HINE for patients in the final efficacy set who did not die or withdraw from the study.
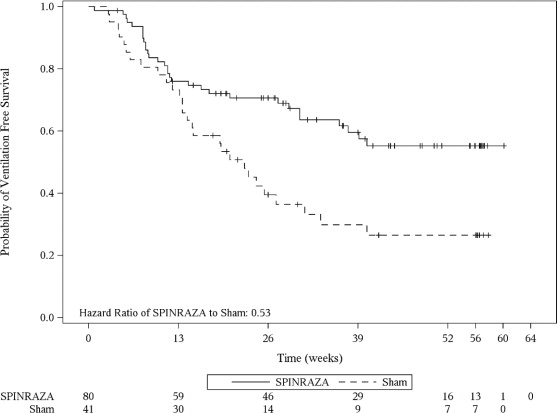
The primary endpoint assessed at the final analysis was time to death or permanent ventilation (≥ 16 hours ventilation/day continuously for > 21 days in the absence of an acute reversible event or tracheostomy). Statistically significant effects on event-free survival and overall survival were observed in patients in the SPINRAZA group compared to those in the sham-control group (Table 4). A 47% reduction in the risk of death or permanent ventilation was observed in the SPINRAZA group (p=0.005) (Figure 2). Median time to death or permanent ventilation was not reached in SPINRAZA group and was 22.6 weeks in the sham-control group. A statistically significant 63% reduction in the risk of death was also observed (p=0.004).
At the final analysis, the study also assessed treatment effects on the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), which is an evaluation of motor skills in patients with infantile-onset SMA. The CHOP-INTEND results are displayed in Table 3.
Table 3. Motor Milestone Response and CHOP-INTEND Results of the Final Analysis of Patients with Infantile-Onset SMA (Study 1) 1At the final analysis, CHOP-INTEND and motor milestone analyses were conducted using the Efficacy Set (SPINRAZA n=73; Sham-control n=37).
2Assessed at the later of Day 183, Day 302, and Day 394 Study Visit
3According to HINE section 2: ≥2 point increase [or maximal score] in ability to kick, OR ≥1 point increase in the motor milestones of head control, rolling, sitting, crawling, standing or walking, AND improvement in more categories of motor milestones than worsening), defined as a responder for this primary analysis.
4Not statistically controlled for multiple comparisons
Endpoint SPINRAZA-treated Patients (n=73) Sham-control Patients (n=37) Motor function Motor milestones1
Proportion achieving pre-defined motor milestone responder criteria (HINE section 2)2,3
37 (51%)
P<0.0001
0 (0%)
CHOP-INTEND1
Proportion achieving a 4-point improvement
Proportion achieving a 4-point worsening4
52 (71%)
p<0.0001
2 (3%)
1 (3%)
17 (46%)Table 4. Survival Results of Patients with Infantile-Onset SMA (Study 1 ) 1At the final analysis, event-free survival and overall survival were assessed using the Intent to Treat population (ITT SPINRAZA n=80; Sham-control n=41).
2Based on log-rank test stratified by disease duration
Endpoint SPINRAZA-treated Patients (n=80) Sham-control Patients (n=41) Survival Event-free survival1
Number of patients who died or received permanent ventilation
Hazard ratio (95% CI)
p-value2
31 (39%)
28 (68%)
0.53 (0.32 -0.89)
p=0.005Overall survival1
Number of patients who died
13 (16%)
16 (39%)
Hazard Ratio (95% CI)
p-value2
0.37 (0.18 – 0.77)
p=0.004Figure 1. Percent of Patients Who Died and Net Change from Baseline in Total Motor Milestone Score (HINE) Among Patients Alive in the Final Efficacy Set of Study 1 *
*For subjects who were alive and ongoing in the study, the change in total motor milestone score was calculated at the later of Day 183, Day 302, or Day 394.
14.2 Later-Onset SMA
Study 2 (NCT02292537) was a multicenter, randomized, double-blind, sham-procedure controlled study in 126 symptomatic children with later-onset SMA (symptom onset after 6 months of age). Patients were randomized 2:1 to either SPINRAZA 12 mg or sham injection as a series of loading doses administered intrathecally followed by maintenance doses administered every 6 months.
The median age at screening was 3 years (range 2-9 years), and the median age of onset of clinical signs and symptoms of SMA was 11 months (range 6-20 months). Of the 126 patients included in the study, 47% were male, 75% were Caucasian, 2% were Black, and 18% were Asian. Length of treatment ranged from 324 to 482 days (median 450 days). At baseline, patients had a mean Hammersmith Functional Motor Scale – Expanded (HFMSE) score of 21.6, all had achieved independent sitting, and no patients had achieved independent walking. Patients in this study were deemed most likely to develop Type 2 or 3 SMA.
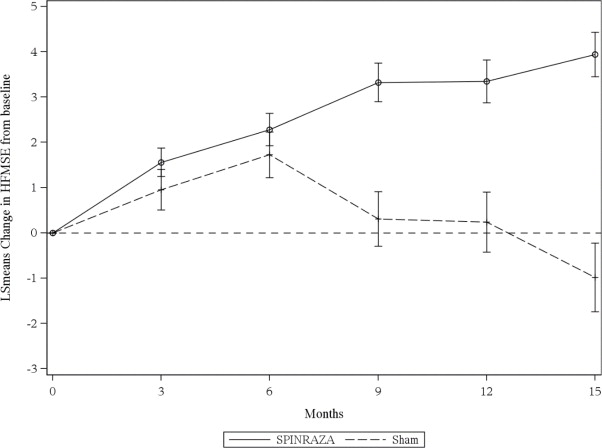
The primary endpoint assessed was the change from baseline score at Month 15 on the HFMSE. The HFMSE evaluates motor function in patients with SMA who have limited ambulation, comprising of 33 scored activities that give objective information on motor ability and clinical progression, such as the ability to sit unassisted, stand, or walk. Each item is scored from 0-2, with a maximum total score of 66. Higher scores indicate better motor function. The primary analysis was conducted in the Intent to Treat (ITT) population, which included all subjects who were randomized and received at least 1 dose of SPINRAZA or at least one sham procedure. At the final analysis, a statistically significant improvement in HFMSE scores from baseline to Month 15 was observed in the SPINRAZA-treated group compared to the sham-control group (Table 5).
Table 5. HFMSE Results in Patients with Later-Onset SMA (Study 2) 1Assessed using the Intent to Treat population who received at least one dose of SPINRAZA or at least one sham procedure (SPINRAZA n=84; Sham-control n=42); data for patients without a Month 15 visit were imputed using the multiple imputation method
2Least squares mean
3Negative value indicates worsening, positive value indicates improvement.
4Based on logistic regression with treatment effect and adjustment for each subject's age at screening and HFMSE score at baseline
Endpoint SPINRAZA-treated Patients (n=84) Sham-control Patients (n=42) HFMSE score
Change from baseline in total HFMSE score at 15 months1,2,3
Proportion of patients who achieved at least a 3-point improvement from baseline to Month 151
3.9 (95% CI: 3.0, 4.9)
p=0.0000001
56.8% (95% CI: 45.6, 68.1)
p=0.00064
-1.0 (95% CI: -2.5, 0.5)
26.3% (95% CI: 12.4, 40.2)
Figure 3. Mean Change from Baseline in HFMSE Score Over Time in the Intent to Treat Set1, 2(Study 2)
1Data for patients without a Month 15 visit were imputed using the multiple imputation method
2Error bars denote +/- standard error
14.3 Presymptomatic SMA
The results of the sham-controlled trial in infantile-onset (Study 1) (NCT02193074) and later-onset (Study 2) (NCT02292537) SMA patients were supported by an open-label uncontrolled trial conducted in 25 presymptomatic SMA patients who had a genetic diagnosis of 5q SMA and 2 or 3 copies of SMN2 (Study 3) (NCT02386553). In Study 3, 15 patients (60%) who had 2 SMN2 copies, and 10 patients (40%) who had 3 SMN2 copies; 48% were male, 56% were Caucasian, 12% were Asian, 4% were American Indian or Alaska Native, and 28% were of another race, or had no race reported. Patients ranged in age from 3 days to 42 days (median 22 days) at the time of first dose. Patients received 12 mg SPINRAZA as a series of loading doses administered intrathecally, followed by maintenance doses administered every 4 months. Patients were assessed with the World Health Organization (WHO) motor milestones, a set of 6 milestones in motor development that would be expected to be attained by 24 months of age in healthy children. An interim analysis was performed after all patients had received SPINRAZA for at least 14 months (median 25 months, range 14 to 34 months). Patients ranged in age from 14 to 34 months (median age of 26 months) at the time of the analysis. At the time of the interim analysis (data cutoff May 2018), all patients receiving SPINRAZA before the onset of SMA symptoms survived without requiring permanent ventilation, and beyond what would be expected based on their SMN2 copy number. All 25 patients (100%) had achieved the WHO motor milestone of sitting without support, and 22 patients (88%) had achieved the milestone of walking with assistance. Of the 22 patients who were older than the age expected to have achieved the ability to walk independently (as defined by the 95th percentile of the WHO expected age of achievement), 17 (77%) achieved the milestone of walking alone (i.e., walking independently).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SPINRAZA injection is a sterile, clear and colorless solution supplied as a 12 mg/5 mL (2.4 mg/mL) solution in a single-dose glass vial free of preservatives. The NDC is 64406-058-01.
16.2 Storage and Handling
Store in a refrigerator between 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze.
SPINRAZA should be protected from light and kept in the original carton until time of use.
If no refrigeration is available, SPINRAZA may be stored in its original carton, protected from light at or below 30oC (86oF) for up to 14 days.
Prior to administration, unopened vials of SPINRAZA can be removed from and returned to the refrigerator, if necessary. If removed from the original carton, the total combined time out of refrigeration should not exceed 30 hours at a temperature that does not exceed 25oC (77oF).
-
17 PATIENT COUNSELING INFORMATION
Thrombocytopenia and Coagulation Abnormalities
Inform patients and caregivers that SPINRAZA could increase the risk of bleeding. Inform patients and caregivers of the importance of obtaining blood laboratory testing at baseline and prior to each dose to monitor for signs of increased potential for bleeding. Instruct patients and caregivers to seek medical attention if unexpected bleeding occurs [see Warnings and Precautions (5.1)].
Renal Toxicity
Inform patients and caregivers that SPINRAZA could cause renal toxicity. Inform patients and caregivers of the importance of obtaining urine testing at baseline and prior to each dose to monitor for signs of potential renal toxicity [see Warnings and Precautions (5.2)].
49655-10
Manufactured for:
Biogen
Cambridge, MA 02142
SPINRAZA is a registered trademark of Biogen.
© Biogen 2016-2024 - PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SPINRAZA
nusinersen injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64406-058 Route of Administration INTRATHECAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Nusinersen (UNII: 5Z9SP3X666) (Nusinersen - UNII:5Z9SP3X666) Nusinersen 2.4 mg in 1 mL Inactive Ingredients Ingredient Name Strength Sodium phosphate, monobasic, dihydrate (UNII: 5QWK665956) 0.05 mg in 1 mL Sodium phosphate, dibasic, anhydrous (UNII: 22ADO53M6F) 0.10 mg in 1 mL Sodium Chloride (UNII: 451W47IQ8X) 8.77 mg in 1 mL Potassium Chloride (UNII: 660YQ98I10) 0.22 mg in 1 mL Calcium chloride (UNII: M4I0D6VV5M) 0.21 mg in 1 mL Magnesium chloride (UNII: 02F3473H9O) 0.16 mg in 1 mL Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64406-058-01 1 in 1 BOX 12/23/2016 1 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209531 12/23/2016 Labeler - Biogen (121376230)