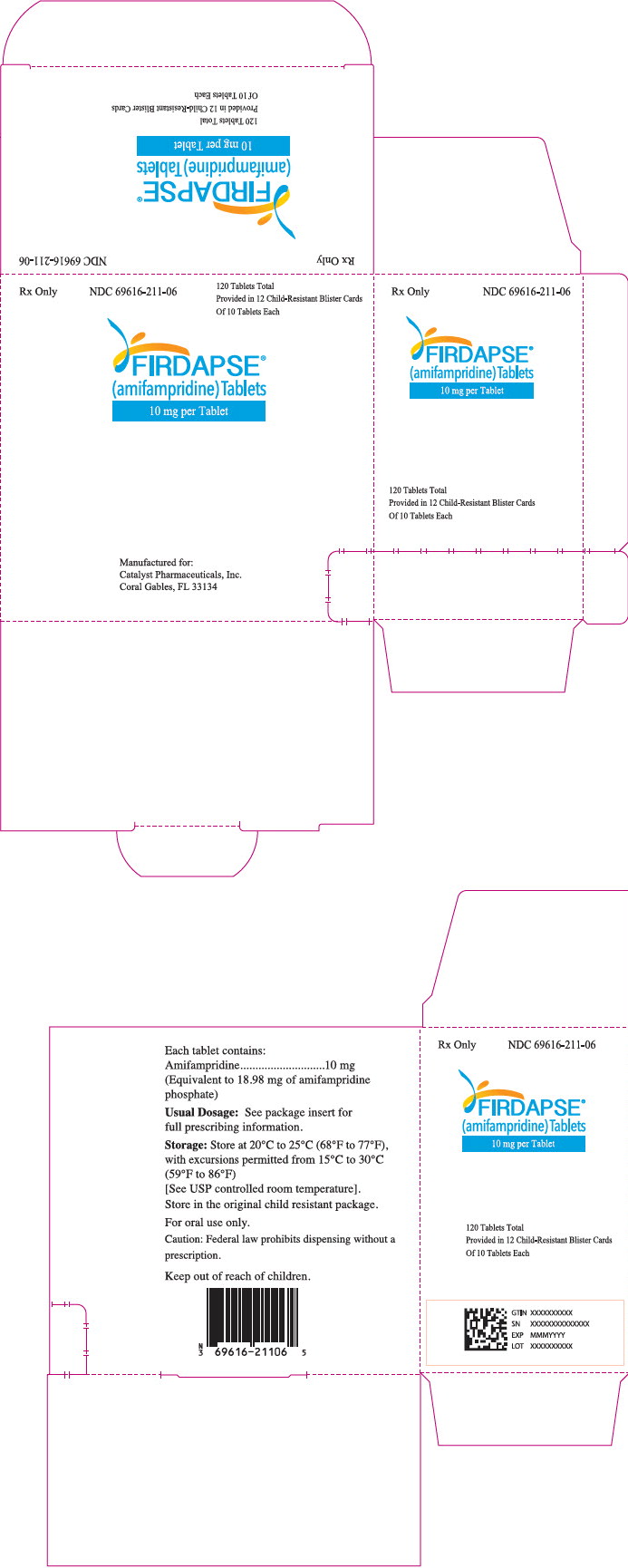
Label: FIRDAPSE- amifampridine phosphate tablet

- NDC Code(s): 69616-211-03, 69616-211-04, 69616-211-06, 69616-211-08
- Packager: Catalyst Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated May 31, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FIRDAPSE® safely and effectively. See full prescribing information for FIRDAPSE®.
FIRDAPSE® (amifampridine) tablets, for oral use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
Dosage and Administration (2.1) 5/2024 INDICATIONS AND USAGE
FIRDAPSE is a potassium channel blocker indicated for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults and pediatric patients 6 years of age and older. (1)
DOSAGE AND ADMINISTRATION
- Administer orally in divided doses (3 to 5 times daily). (2.1)
- The recommended starting dosage for adults (any weight) and pediatric patients weighing 45 kg or more is 15 mg to 30 mg daily, in divided doses. (2.1)
- The recommended starting dosage for pediatric patients weighing less than 45 kg is 5 mg to 15 mg daily, in divided doses. (2.1)
- The recommended starting dosage for adult and pediatric patients with renal impairment, hepatic impairment, and in known N-acetyltransferase 2 (NAT2) poor metabolizers is the lowest recommended initial daily dosage (2.1, 2.2, 2.3, 2.4)
- For patients with a dosage adjustment of less than 5 mg increments, or who have difficulty swallowing, or require feeding tube, a l mg/mL suspension can be prepared. (2.5)
DOSAGE FORMS AND STRENGTHS
- Tablets: 10 mg, functionally scored. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Seizures: FIRDAPSE can cause seizures. Consider discontinuation or dose-reduction of FIRDAPSE in patients who have a seizure while on treatment. (5.1)
- Hypersensitivity reactions: If a hypersensitivity reaction such as anaphylaxis occurs, FIRDAPSE should be discontinued and appropriate therapy initiated. (5.2)
ADVERSE REACTIONS
The most common (> 10%) adverse reactions are: paresthesia, upper respiratory tract infection, abdominal pain, nausea, diarrhea, headache, elevated liver enzymes, back pain, hypertension, and muscle spasms. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Catalyst Pharmaceuticals at 1-844-347-3277 (1-844-FIRDAPSE) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Drugs that lower seizure threshold: Concomitant use may lead to an increased risk of seizures. (7.1)
- Drugs with cholinergic effects (e.g., direct or indirect cholinesterase inhibitors): Concomitant use may increase the cholinergic effects of FIRDAPSE and of those drugs and increase the risk of adverse reactions. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
2.2 Patients with Renal Impairment
2.3 Patients with Hepatic Impairment
2.4 Known N-acetyltransferase 2 (NAT2) Poor Metabolizers
2.5 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Seizures
5.2 Hypersensitivity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Drugs that Lower Seizure Threshold
7.2 Drugs with Cholinergic Effects
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 NAT2 Poor Metabolizers
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended oral dosage of FIRDAPSE for adults and pediatric patients 6 years of age and older is included in Table 1. For pediatric patients, the recommended dosing regimen is dependent on body weight. Dosage should be increased every 3 to 4 days based on clinical response and tolerability. Titration increments should not exceed those shown in Table 1.
Table 1: Recommended Oral Dosage for the Treatment of LEMS in Adults and Pediatric Patients 6 Years of Age and Older *See Dosage and Administration (2.5) for a method to achieve these doses
Age and Body Weight Initial Daily Dosage* Titration Regimen Maximum Single
DoseMaximum Total Daily
Maintenance Dosage- Adults (any weight)
- Pediatric patients weighing 45 kg or more
15 mg to 30 mg daily,
in 3 to 5 divided dosesIncrease total daily dosage
by 5 mg
every 3 or 4 days20 mg 100 mg
Given in divided doses- Pediatric patients weighing less than 45 kg
5 mg to 15 mg daily,
in 3 to 5 divided dosesIncrease total daily dosage by 2.5 mg every 3 or 4 days 10 mg 50 mg
Given in divided doses2.2 Patients with Renal Impairment
The recommended starting dosage of FIRDAPSE in patients with renal impairment [creatinine clearance (CLcr) 15 to 90 mL/min] is the lowest recommended initial daily dosage (i.e., 15 mg daily for pediatric patients weighing 45 kg or more and for adults, and 5 mg daily for pediatric patients weighing less than 45 kg) taken orally in divided doses. No dosage recommendation for FIRDAPSE can be made for patients with end-stage renal disease [see Dosage and Administration (2.1), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
2.3 Patients with Hepatic Impairment
The recommended starting dosage of FIRDAPSE in patients with any degree of hepatic impairment is the lowest recommended initial daily dosage (i.e., 15 mg daily for pediatric patients weighing 45 kg or more and for adults, and 5 mg daily for pediatric patients weighting less than 45 kg) taken orally in divided doses [see Dosage and Administration (2.1), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
2.4 Known N-acetyltransferase 2 (NAT2) Poor Metabolizers
The recommended starting dosage of FIRDAPSE in known N-acetyltransferase 2 (NAT2) poor metabolizers is the lowest recommended initial daily dosage (i.e., 15 mg daily for pediatric patients weighing 45 kg or more and for adults, and 5 mg daily for pediatric patients weighing less than 45 kg) taken orally in divided doses [see Dosage and Administration (2.1), Use in Specific Populations (8.8), and Clinical Pharmacology (12.5)].
2.5 Administration Instructions
FIRDAPSE can be taken without regard to food.
Preparation of a 1mg/mL Suspension (see the Instructions for Use for full instructions on how to prepare the 1mg/mL suspension)
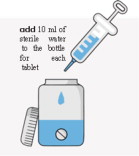
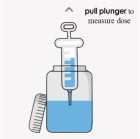
When patients require a dosage in less than 5 mg increments, have difficulty swallowing tablets, or require feeding tubes, a 1 mg/mL suspension can be prepared (e.g., by placing the required number of tablets in a 50 to 100 mL container, adding 10 mL of sterile water for each tablet, waiting for 5 minutes, and shaking well for 30 seconds).
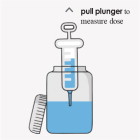
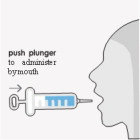
Crushing the tablets prior to making the suspension is not necessary. After preparation of the suspension, an oral syringe can be used to draw up and administer the correct dose by mouth or by feeding tube.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Seizures
FIRDAPSE can cause seizures. Seizures have been observed in patients without a history of seizures taking FIRDAPSE at the recommended doses, at various times after initiation of treatment, at an incidence of approximately 2%. Many of the patients were taking medications or had comorbid medical conditions that may have lowered the seizure threshold [see Drug Interactions (7.1)]. Seizures may be dose-dependent. Consider discontinuation or dose-reduction of FIRDAPSE in patients who have a seizure while on treatment. FIRDAPSE is contraindicated in patients with a history of seizures.
5.2 Hypersensitivity
In clinical trials, hypersensitivity reactions and anaphylaxis associated with FIRDAPSE administration have not been reported. Anaphylaxis has been reported in patients taking another aminopyridine; therefore, it may occur with FIRDAPSE. If anaphylaxis occurs, administration of FIRDAPSE should be discontinued and appropriate therapy initiated.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
6.1 Clinical Trials Experience
Adults
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled and uncontrolled clinical trials (Study 1 and 2) in patients with LEMS [see Clinical Studies (14)], 63 patients were treated with FIRDAPSE, including 40 patients treated for more than 6 months, and 39 patients treated for more than 12 months. In an expanded access program, 139 patients with LEMS were treated with FIRDAPSE, including 102 patients treated for more than 6 months, 77 patients treated for more than 12 months, and 53 patients treated for more than 18 months. In the expanded access program, patients were treated with FIRDAPSE with up to 100 mg daily in divided doses.
Study 1 was a double-blind, placebo-controlled, randomized discontinuation study in adults with LEMS. Following an initial open- label run-in phase (up to 90 days), patients were randomized to either continue FIRDAPSE treatment or transition to placebo, for a 14-day double-blind phase. Following final assessments, patients were allowed to resume FIRDAPSE treatment for up to 2 years (open-label long-term safety phase of the study).
During the open-label run-in phase of Study 1, 53 patients received FIRDAPSE for an average of 81 days at a mean daily dosage of 50.5 mg/day. The mean patient age was 52.1 years and 66% were female. There were 42 patients who had no prior exposure to FIRDAPSE at the initiation of this study. Table 2 shows adverse reactions with an incidence of 5% or greater occurring in the 42 LEMS patients newly initiated on treatment with FIRDAPSE during the run-in phase of the study.
Table 2. Adverse Reactions in ≥5% of LEMS Patients Newly Treated with FIRDAPSE in Study 1 *Includes paresthesia, oral paresthesia, oral hypoesthesia
**Includes elevated alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and gamma-glutamyl transferase (GGT)
Adverse Reaction FIRDAPSE
N=42
%Paresthesia* 62 Upper respiratory tract infection 33 Abdominal pain 14 Nausea 14 Diarrhea 14 Headache 14 Elevated liver enzymes** 14 Back pain 14 Hypertension 12 Muscle spasms 12 Dizziness 10 Asthenia 10 Muscular weakness 10 Pain in extremity 10 Cataract 10 Constipation 7 Bronchitis 7 Fall 7 Lymphadenopathy 7 Other Adverse Reactions
In the overall population treated in Study 1 (n=53), including the double-blind phase and the 2-year open-label long-term safety phase, additional adverse reactions occurring in at least 5% of the patients included: dyspnea, urinary tract infection, gastroesophageal reflux, insomnia, peripheral edema, pyrexia, viral infection, blood creatine phosphokinase increase, depression, erythema, hypercholesterolemia, and influenza. These patients received a mean daily dosage of 66 mg of FIRDAPSE.
Pediatrics
Safety of FIRDAPSE was evaluated in pediatric patients in an expanded access program, where 21 pediatric patients received FIRDAPSE for at least 1 year. Adverse reactions reported in pediatric patients were similar to those seen in adult patients, with the exception of clinically significant weight loss in two pediatric patients at doses of 60 mg per day and higher.
-
7 DRUG INTERACTIONS
7.1 Drugs that Lower Seizure Threshold
The concomitant use of FIRDAPSE and drugs that lower seizure threshold may lead to an increased risk of seizures [see Warnings and Precautions (5.1)]. The decision to administer FIRDAPSE concomitantly with drugs that lower the seizure threshold should be carefully considered in light of the severity of the associated risks.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to FIRDAPSE during pregnancy. Physicians are encouraged to enroll pregnant patients, or pregnant women may register themselves in the registry by calling 855- 212-5856 (toll-free), using the Fax number 877-867-1874 (toll-free), by contacting the Pregnancy Coordinating Center at firdapsepregnancyregistry@ubc.com, or by visiting the study website www.firdapsepregnancystudy.com.
Risk Summary
There are no data on the developmental risk associated with the use of FIRDAPSE in pregnant women. In animal studies, administration of amifampridine phosphate to rats during pregnancy and lactation resulted in developmental toxicity (increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development) at doses associated with maternal plasma drug levels lower than therapeutic drug levels (see Animal Data). In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of amifampridine phosphate (0, 7.5, 22.5, or 75 mg/kg/day) to female rats prior to and during mating and continuing throughout organogenesis produced no adverse effects on embryofetal development. The highest dose tested is approximately 4 times the maximum recommended human dose (MRHD) of amifampridine (100 mg/day) on a body surface area (mg/m2) basis. Oral administration of amifampridine phosphate (0, 9, 30, or 57 mg/kg/day) to pregnant rabbits throughout organogenesis produced no adverse effects on embryofetal development. The highest dose tested is approximately 6 times the MRHD of amifampridine on a body surface area (mg/m2) basis.
Oral administration of amifampridine phosphate (0, 7.5, 22.5, or 75 mg/kg/day) to female rats throughout pregnancy and lactation resulted in an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups at the mid and high doses tested. The no-effect dose (7.5 mg/kg/day amifampridine phosphate) for adverse developmental effects is associated with a plasma amifampridine exposure (AUC) less than that in humans at the MRHD of amifampridine.
8.2 Lactation
Risk Summary
There are no data on the presence of FIRDAPSE in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for FIRDAPSE and any potential adverse effects on the breastfed infant from FIRDAPSE or from the underlying maternal condition.
In lactating rat, amifampridine was excreted in milk and reached levels similar to those in maternal plasma.
8.4 Pediatric Use
Safety and effectiveness of FIRDAPSE for the treatment of LEMS have been established in pediatric patients 6 years of age and older. Use of FIRDAPSE for this indication is supported by evidence from adequate and well-controlled studies of FIRDAPSE in adults with LEMS, pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients, and safety data from pediatric patients aged 6 years and older [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14)].
Safety and effectiveness in pediatric patients below the age of 6 years have not been established.
8.5 Geriatric Use
Clinical studies of FIRDAPSE did not include a sufficient number of subjects aged 65 and over (19 of 63 patients in Studies 1 and 2) to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Dosage and Administration (2.2, 2.3) and Drug Interactions (7.1, 7.2)].
8.6 Renal Impairment
Renal clearance is an elimination pathway for amifampridine and the inactive metabolite, 3-N-acetyl-amifampridine, and exposure of amifampridine is higher in subjects with renal impairment [see Clinical Pharmacology (12.3)]. Therefore, in patients with renal impairment, FIRDAPSE should be initiated at the lowest recommended initial daily dosage, and patients should be closely monitored for adverse reactions [see Dosage and Administration (2.2)]. Consider dosage modification or discontinuation of FIRDAPSE for patients with renal impairment as needed based on clinical effect and tolerability. The safety, efficacy, and pharmacokinetics of amifampridine have not been studied in patients with end-stage renal disease (CLcr <15 mL/min or patients requiring dialysis). No dosage recommendation for FIRDAPSE can be made for patients with end-stage renal disease.
8.7 Hepatic Impairment
In patients with any degree of hepatic impairment, FIRDAPSE should be initiated at the lowest recommended initial daily dosage, and patients should be monitored for adverse reactions [see Dosage and Administration (2.3)]. Consider dosage modification or discontinuation of FIRDAPSE for patients with hepatic impairment as needed based on clinical effect and tolerability.
8.8 NAT2 Poor Metabolizers
Exposure of FIRDAPSE is increased in patients who are N-acetyltransferase 2 (NAT2) poor metabolizers [see Clinical Pharmacology (12.5)]. Therefore, initiate FIRDAPSE in patients who are known NAT2 poor metabolizers at the lowest recommended initial daily dosage and monitor for adverse reactions [see Dosage and Administration (2.4)]. Consider dosage modification of FIRDAPSE for patients who are known NAT2 poor metabolizers as needed based on clinical effect and tolerability.
-
10 OVERDOSAGE
Overdose with FIRDAPSE was not reported during clinical studies.
In a case report, a 65-year-old patient with LEMS inadvertently received a total daily amifampridine dose of 360 mg/day (approximately 4 times the maximum recommended total daily dose) and was hospitalized for general weakness, paresthesia, nausea, vomiting, and palpitations. The patient developed convulsions and paroxysmal supraventricular tachycardia, and four days after admission, experienced cardiac arrest. The patient was resuscitated and ultimately recovered following withdrawal of amifampridine.
Patients with suspected overdose with FIRDAPSE should be monitored for signs or symptoms of exaggerated FIRDAPSE adverse reactions or effects, and appropriate symptomatic treatment instituted immediately.
-
11 DESCRIPTION
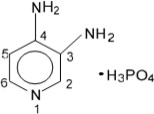
The active ingredient of FIRDAPSE is amifampridine phosphate, which is a voltage-gated potassium channel blocker. Amifampridine phosphate is described chemically as 3,4-diaminopyridine phosphate with a molecular weight of 207.1 and a molecular formula of C5H7N3 • H3PO4. The structural formula is:
Amifampridine phosphate is a white, crystalline powder that is freely soluble in water, and slightly soluble in solvents ethanol, methanol and acetic acid. A 1% aqueous solution of amifampridine phosphate has a pH of 4.4 at ambient conditions.
Each FIRDAPSE tablet contains 10 mg amifampridine (equivalent to 18.98 mg amifampridine phosphate). The tablet formulation includes the following inactive ingredients: calcium stearate, colloidal silicon dioxide, and microcrystalline cellulose.
FIRDAPSE tablets are intended for oral administration only.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which amifampridine exerts its therapeutic effect in LEMS patients has not been fully elucidated. Amifampridine is a broad-spectrum potassium channel blocker.
12.2 Pharmacodynamics
The effect of FIRDAPSE on QTc interval prolongation was studied in a double blind, randomized, placebo and positive controlled study in 52 healthy individuals who are slow acetylators. At an exposure 2-fold the expected maximum therapeutic exposure of amifampridine, FIRDAPSE did not prolong QTc to any clinically relevant extent.
12.3 Pharmacokinetics
The pharmacokinetics of amifampridine are similar between healthy individuals and LEMS patients. Following single and multiple doses, AUC, Cmax and Cmin were highly variable between individuals. FIRDAPSE exposure increased proportionally with dose across the range of 20 mg to 80 mg single oral doses.
Absorption
Amifampridine peak plasma concentration is reached 20 minutes to 1 hour after administration. Food does not have a clinically significant effect on the exposure of amifampridine.
Elimination
Amifampridine is eliminated primarily through metabolism to 3-N-acetyl-amifampridine and to a smaller extent through the kidneys. The terminal half-life ranges from 1.8 to 2.5 hours in healthy subjects.
Specific Populations
Pediatric Patients (6 Years of Age and Older)
A population pharmacokinetic analysis showed that body weight significantly correlates with the clearance of amifampridine; clearance increased with an increase in body weight. A weight-based dosing regimen is necessary to achieve amifampridine exposures in pediatric patients 6 years of age and older, similar to the exposures observed in adults at recommended doses of FIRDAPSE [see Dosage and Administration (2.1) and Clinical Studies (14)].
Patients with Renal Impairment
Pharmacokinetic data are available from a study of 24 otherwise healthy subjects with impaired renal function who received a single 10-mg dose of FIRDAPSE. The exposure of amifampridine (measured as AUC) was 2- to 3-fold higher in subjects with moderate (CLcr 30-59 mL/min) or severe (CLcr 15-29 mL/min) renal impairment than in subjects with normal renal function (CLcr greater than or equal to 90 mL/min). Compared with subjects with normal renal function, subjects with mild renal impairment (CLcr 60-89 mL/min) had a 36% increase in exposure. Therefore, FIRDAPSE should be initiated at the lowest recommended initial daily dosage in patients with renal impairment, and such patients should be closely monitored for adverse reactions [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)]. Cmax was marginally affected by renal impairment.
Patients with Hepatic Impairment
A study was conducted to determine the PK, safety, and tolerability of FIRDAPSE (amifampridine phosphate) administered at 10 mg to subjects with moderate hepatic impairment (N = 7) compared to demographically matched healthy subjects with normal hepatic function (N = 9). The subjects with moderate hepatic impairment had a 22% higher Cmax and a 33% higher AUCinf for amifampridine than subjects with normal hepatic function. FIRDAPSE was not evaluated in subjects with severe hepatic impairment [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
12.5 Pharmacogenomics
Genetic variants in the N-acetyltransferase gene 2 (NAT2) affect the rate and extent of FIRDAPSE metabolism. Poor metabolizers, also referred to as “slow acetylators” (i.e., carriers of two reduced function alleles), have 3.5- to 4.5-fold higher Cmax, and 5.6- to 9- fold higher AUC than normal metabolizers, also referred to as “fast/rapid acetylators” (i.e., carriers of two normal function alleles). Therefore, FIRDAPSE should be initiated at the lowest recommended initial daily dosage in known NAT2 poor metabolizers, and such patients should be closely monitored for adverse reactions [see Dosage and Administration (2.4) and Use in Specific Populations (8.8)]. In the general population, the NAT2 poor metabolizer phenotype prevalence is 40–60% in the White and African American populations, and in 10–30% in Asian ethnic populations (individuals of Japanese, Chinese, or Korean descent).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
In a 104-week carcinogenicity study in rats, oral administration of amifampridine phosphate (0, 15, 48, or 105 mg/kg/day) resulted in an increase in uterine tumors (endometrial carcinoma and combined endometrial adenoma/endometrial carcinoma/squamous cell carcinoma) at the mid and high doses tested. The low dose, not associated with an increase in tumors, is less than the maximum recommended human dose (MRHD) of amifampridine (100 mg/day) on a body surface area (mg/m2 basis).
In a 26-week carcinogenicity study in transgenic (Tg.rasH2) mice, oral administration of amifampridine phosphate (0, 2, 7, or 21 mg/kg/day in males and 0, 3, 10, or 30 mg/kg/day in females) resulted in no increase in tumors.
Mutagenesis
Amifampridine phosphate was negative in the in vitro bacterial reverse mutation and in vivo rat micronucleus assays. Amifampridine phosphate was positive for clastogenicity in the in vitro mouse lymphoma tk assay in the absence of metabolic activation.
Impairment of Fertility
Oral administration of amifampridine phosphate (0, 7.5, 22.5, or 75 mg/kg/day) to male and female rats prior to and during mating, and continuing in females throughout organogenesis, produced no adverse effects on fertility. The highest dose tested is approximately 4 times the MRHD of amifampridine on a body surface area (mg/m2) basis.
-
14 CLINICAL STUDIES
The efficacy of FIRDAPSE for the treatment of LEMS was demonstrated in two randomized, double-blind, placebo-controlled discontinuation studies. A total of 64 adults (age 21 to 88 years) with LEMS were enrolled (Study 1 and Study 2). The studies enrolled patients with a confirmed diagnosis of LEMS based on either neurophysiology studies or a positive anti-P/Q type voltage-gated calcium channel antibody test. Patients were required to be on an adequate and stable dosage (30 to 80 mg daily) of amifampridine phosphate prior to entering the randomized discontinuation phases of both studies.
The two co-primary measures of efficacy in both studies were the change from baseline to the end of the discontinuation period in the Quantitative Myasthenia Gravis (QMG) score and in the Subject Global Impression (SGI) score.
The QMG is a 13-item physician-rated categorical scale assessing muscle weakness. Each item is assessed on a 4-point scale, where a score of 0 represents no weakness, and a score of 3 represents severe weakness (total score 0-39). Higher scores represent greater impairment.
The SGI is a 7-point scale on which patients rated their global impression of the effects of the study treatment on their physical well- being. Lower scores on the SGI represent lower perceived benefit with the study treatment.
A key secondary efficacy endpoint was the clinical global impression improvement (CGI-I) score, a 7-point scale on which the treating physician rated the global impression of change in clinical symptoms. A higher CGI-I score indicates a perceived worsening of clinical symptoms.
Study 1 (NCT01377922)
After an initial open-label run-in phase, 38 patients were randomized in a double-blind fashion to either continue treatment with FIRDAPSE (n=16) or to a downward titration to placebo (n=22) over 7 days. Following the downward titration period, patients remained on blinded FIRDAPSE or placebo for 7 more days. Efficacy was assessed at Day 14 of the double-blind period. Patients were allowed to use stable dosages of peripherally acting cholinesterase inhibitors or oral immunosuppressants. Twenty-six percent of patients randomized to FIRDAPSE were receiving cholinesterase inhibitors, versus 36% in the placebo group, and 28% of patients randomized to FIRDAPSE were receiving oral immunosuppressant therapies, versus 34% in the placebo group.
Patients had a median age of 54 years (range: 21 to 88 years), 61% were female, and 90% were White. Eighty-four percent of patients had a diagnosis of autoimmune LEMS, and 16% of patients had a diagnosis of paraneoplastic LEMS.
During the double-blind period (from Baseline to Day 14), the QMG scores tended to worsen in both treatment groups, but there was significantly greater worsening in the placebo group than in the FIRDAPSE group (p=0.045). Similarly, the SGI score tended to worsen in both treatment groups during the double-blind period, but there was significantly greater worsening in the placebo group than in the FIRDAPSE group (p=0.003), as summarized in Table 3. These results indicate that in Study 1, patients randomized to placebo had a significantly greater worsening of muscle weakness and of global impression of the effects of the study treatment on their physical well-being, compared to patients who continued FIRDAPSE in the double-blind period.
Table 3. Change from Baseline to Day 14 in QMG Score and SGI Score in Study 1 a. QMG Score range 0 (no impairment) to 39 (worst impairment)
b. SGI Score range 0 (least perceived benefit) to 7 (most perceived benefit)
c. Pairwise contrast at Day 14 from mixed-effects model with repeated measures.
Assessment FIRDAPSE (n=16) Placebo (n=21) Primary Endpoints QMG Scorea Baseline (mean) 6.4 5.6 Change from Baseline (Least Square Mean) 0.4 2.2 FIRDAPSE-placebo Treatment Difference (Least Square Mean (95% CI))
–1.7 (–3.4, –0.0)p-valuec 0.045 SGI Scoreb Baseline (mean) 5.6 5.9 Change from Baseline (Least Square Mean) -0.8 -2.6 FIRDAPSE-placebo Treatment Difference, (Least Square Mean (95% CI))
1.8 (0.7, 3.0)p-valuec 0.003 The CGI-I score was significantly greater for patients randomized to placebo than for patients who continued treatment with FIRDAPSE, with a mean difference between FIRDAPSE and placebo of -1.1 (p=0.02), indicating that clinicians perceived a greater worsening of clinical symptoms in patients who were randomized to placebo and discontinued from FIRDAPSE treatment, compared to patients who continued FIRDAPSE in the double-blind period.
Study 2 (NCT02970162)
Patients on stable treatment with FIRDAPSE were randomized 1:1 in a double-blind fashion to either continue treatment with FIRDAPSE (n=13) or change to placebo (n=13) for 4 days. Efficacy was assessed at the end of the 4-day double-blind discontinuation period. Patients were allowed to use stable doses of peripherally acting cholinesterase inhibitors or corticosteroids. Sixty-one percent of patients randomized to FIRDAPSE were receiving cholinesterase inhibitors, versus 54% of patients randomized to placebo.
Corticosteroid use was similar between FIRDAPSE and placebo (8%). Patients with recent use of immunomodulatory therapies (e.g., azathioprine, mycophenolate, cyclosporine), rituximab, intravenous immunoglobulin G, and plasmapheresis were excluded from the study. Patients had a median age of 55.5 years (range: 31 to 75 years), 62% were female, and 88% were white.
From Baseline to Day 4, there was significantly greater worsening in the QMG score in the placebo group than in the FIRDAPSE group (p=0.0004), and also significantly greater worsening in the SGI score in the placebo group than in the FIRDAPSE group (p=0.0003), as summarized in Table 4. These results indicate that in Study 2, patients randomized to placebo had a significantly greater worsening of muscle weakness and of global impression of the effects of the study treatment on their physical well-being, compared to patients who continued FIRDAPSE in the double-blind period.
Table 4. Change from Baseline to Day 4 in QMG Scores and SGI Scores in Study 2 Assessment FIRDAPSE (n=13) Placebo (n=13) a. QMG Score range 0 (no impairment) to 39 (worst impairment)
b. SGI Score range 0 (least perceived benefit) to 7 (most perceived benefit)
c. Change from baseline for QMG total score was modeled as the response, with fixed effects terms for treatment and QMG at Baseline
d. p-value based on the Wilcoxon Rank Sum Test for treatment differences.
QMG Scoresa Baseline, Mean 7.8 8.5
Change from Baseline, Least Square Meanc
0.00
6.54FIRDAPSE-placebo Treatment Difference, Least Square Mean (95% CI)
-6.54 (-9.78, -3.29)p-valued 0.0004 SGI Scoresb Baseline, Mean 6.1 5.8
Change from Baseline, Least Square Meanc
-0.64
-3.59FIRDAPSE-placebo Treatment Difference,
Least Square Mean (95% CI)
2.95 (1.53, 4.38)p-valued 0.0003 The clinical global impression improvement (CGI-I) score was significantly greater for patients randomized to placebo than for patients who continued treatment with FIRDAPSE, with a mean difference between FIRDAPSE and placebo of -2.7 (p=0.002), indicating that clinicians perceived a greater worsening of clinical symptoms in patients who were randomized to placebo and discontinued from FIRDAPSE treatment, compared to patients who continued FIRDAPSE in the double-blind period.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
FIRDAPSE 10 mg tablets are white to off white-round, and functionally scored. Each tablet is debossed on the non-scored side with “CATALYST” and on the scored side with “211” above the score and “10” below the score. Tablets can be divided in half at the score. FIRDAPSE is supplied as follows:
16.2 Storage and Handling
Store FIRDAPSE tablets at 20°C to 25°C (68°F to 77°F) with excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP controlled room temperature]. See Dosage and Administration (2.5) for storage instructions for FIRDAPSE prepared suspension.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Risk of Seizures
Inform patients that FIRDAPSE can cause seizures, and to notify their healthcare provider if they experience a seizure [see Warnings and Precautions (5.1)].
Hypersensitivity
Instruct patients to inform their healthcare provider if they have signs or symptoms of hypersensitivity, and to seek emergency help if symptoms of anaphylaxis occur [see Warnings and Precautions (5.2)].
FIRDAPSE Dosing
Instruct patients to take FIRDAPSE exactly as prescribed. Patients should carefully follow the dose escalation schedule provided by their healthcare provider to safely achieve the therapeutic dosage [see Dosage and Administration (2)]. Inform patients that the tablets may be divided in half at the score, if needed. Instruct patients not to take a double dose to make up for a missed dose.
If they require a dosage in less than 5 mg increments, have difficulty swallowing tablets, or require feeding tubes, refer patients and/or caregivers to the Instructions for Use on how to prepare a 1 mg/mL suspension [see Dosage and Administration (2.5)]. If the patient requires treatment with the 1 mg/mL FIRDAPSE suspension, advise patients and/or caregivers that supplies required to prepare the suspension may be obtained at their local pharmacy.
Drug Interactions
Instruct patients to notify their healthcare provider prior to starting any new medication, including over-the-counter drugs [see Drug Interactions (7)].
Pregnancy
Instruct patients that if they are pregnant or plan to become pregnant while taking FIRDAPSE they should inform their healthcare provider. Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to FIRDAPSE during pregnancy and encourage them to enroll if they become pregnant while taking FIRDAPSE [see Use in Specific Populations (8.1)].
Storage
Advise patients to store FIRDAPSE at 68ºF to 77ºF (20ºC to 25ºC).
Instruct patients and/or caregivers who prepare the 1 mg/mL suspension of FIRDAPSE that it should be prepared daily and refrigerated between doses. The suspension can be stored under refrigeration for up to 24 hours. Instruct the patient and/or caregiver to discard any unused portion of the suspension after 24 hours.
Distributed by Catalyst Pharmaceuticals, Inc., Coral Gables, FL 33134
FIRDAPSE is a trademark of SERB S.A.
Catalyst and the Catalyst logo are trademarks of Catalyst Pharmaceuticals, Inc.© 2024 Catalyst Pharmaceuticals, Inc. – All rights reserved.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration
Revised: 5/2024
MEDICATION GUIDE
FIRDAPSE® (FIR-dapse)
(amifampridine)
tablets, for oral useRead this Medication Guide before you start taking FIRDAPSE and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. What is the most important information I should know about FIRDAPSE?
FIRDAPSE can cause seizures.
- You could have a seizure even if you never had a seizure before.
- Do not take FIRDAPSE if you have ever had a seizure.
Stop taking FIRDAPSE and call your doctor right away if you have a seizure while taking FIRDAPSE. What is FIRDAPSE?
FIRDAPSE is a prescription medicine used to treat Lambert-Eaton myasthenic syndrome (LEMS) in people 6 years of
age and older.
It is not known if FIRDAPSE is safe or effective in children less than 6 years of age.Do not take FIRDAPSE if you:
- have ever had a seizure.
- are allergic to amifampridine phosphate, or another aminopyridine.
Before you take FIRDAPSE, tell your doctor about all of your medical conditions. including if you:
- are taking another aminopyridine, such as compounded 3,4-diaminopyridine (3,4-DAP)
- have had a seizure
- have kidney problems
- have liver problems
- are pregnant or plan to become pregnant. It is not known if FIRDAPSE will harm your unborn baby. You and your doctor will decide if you should take FIRDAPSE while you are pregnant.
- There is a registry for women who become pregnant during treatment with FIRDAPSE. The purpose of this registry is to collect information about your health and your baby's health. Contact the registry as soon as you learn that you are pregnant, or ask your doctor to contact for you by calling 855-212-5856 (toll free), contacting the Fax number 877-867-1874 (toll free), emailing the Pregnancy Coordinating Center at firdapsepregnancyregistry@ubc.com, or visiting the study website www.firdapsepregnancystudy.com
- are breastfeeding or plan to breastfeed. It is not known if FIRDAPSE passes into your breast milk. Talk to your doctor about the best way to feed your baby while taking FIRDAPSE.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. How should I take FIRDAPSE?
- If your dose is less than 5mg, you have trouble swallowing tablets, or a feeding tube is needed, see the detailed Instructions for Use on how to take and prepare a suspension of FIRDAPSE.
- Take FIRDAPSE exactly as your doctor tells you to take it.
- Do not change your dose of FIRDAPSE.
- Do not stop taking FIRDAPSE without first talking to your doctor.
- FIRDAPSE tablets are scored and can be split if less than a full tablet is needed for you to get the right dose.
- FIRDAPSE can be taken with or without food.
- If you miss a dose of FIRDAPSE, skip that dose and take your next dose at your next scheduled dose time. Do not double your dose to make up the missed dose.
- Do not take FIRDAPSE together with other medicines known to increase the risk of seizures.
- If you take too much FIRDAPSE, call your doctor or go to the nearest hospital emergency room right away.
What are the possible side effects of FIRDAPSE?
FIRDAPSE may cause serious side effects, including:
- Seizures. See “What is the most important information I should know about FIRDAPSE?”
-
Serious allergic reactions, such as anaphylaxis. FIRDAPSE can cause serious allergic reactions. Stop taking FIRDAPSE and call your doctor right away or get emergency medical help if you have:
- shortness of breath or trouble breathing
- swelling of your throat or tongue
- hives
The most common side effects of FIRDAPSE include:
- tingling around the mouth, tongue, face, fingers, toes, and other body parts
- upper respiratory infection
- stomach pain
- nausea
- diarrhea
- headache
- increased liver enzymes
- back pain
- high blood pressure
- muscle spasms
Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of FIRDAPSE.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store FIRDAPSE?
- Store FIRDAPSE tablets at room temperature between 68ºF to 77ºF (20ºC to 25ºC).
- Safely throw away FIRDAPSE tablets that are out of date or no longer needed.
- Store FIRDAPSE prepared oral suspension in the refrigerator between 36°F to 46°F (2°C to 8°C) between doses for up to 24 hours.
- Safely throw away unused FIRDAPSE oral suspension after 24 hours.
Keep FIRDAPSE and all medicines out of the reach of children. General Information about the safe and effective use of FIRDAPSE
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FIRDAPSE for a condition for which it was not prescribed. Do not give FIRDAPSE to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk to your doctor or pharmacist. You can ask your pharmacist or doctor for information about FIRDAPSE that is written for health professionals.What are the ingredients in FIRDAPSE?
Active ingredient: amifampridine
Inactive ingredients: calcium stearate, colloidal silicon dioxide, and microcrystalline cellulose.
Distributed by Catalyst Pharmaceuticals, Inc., Coral Gables, FL 33134
For more information, go to www.YourCatalystPathways.com or call 1-833-422-8259 -
INSTRUCTIONS FOR USE
Instructions for Use
FIRDAPSE (FIR-dapse)
(amifampridine) tablets
for oral use
This Instructions for Use contains information on how to mix and use FIRDAPSE prepared suspension. The FIRDAPSE prepared suspension can be used for people who are prescribed a dosage that cannot be obtained with whole or half tablets, who have trouble swallowing tablets, or who have a feeding tube.
Prepare FIRDAPSE oral suspension each day.
Supplies you will need:
You can get these supplies at your local pharmacy.


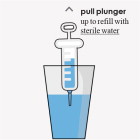
your FIRDAPSE tablets empty bottle with cap (50-100 mL recommended) sterile water oral syringe with catheter tip (10mL, may require smaller syringe for dosing) Instructions to make a 1 mg/mL suspension:
Important:
- Do not use any foods or liquids other than sterile water to mix FIRDAPSE.
- You can prepare each dose separately or all of your doses for the day at one time.
Instructions to administer the prepared suspension:
Important:
- Prepare fresh suspensions daily.
- If you prepare all of your doses for the day at one time, refrigerate the suspension between doses. Shake well before drawing up each dose.
OPTION 1 To administer by mouth:
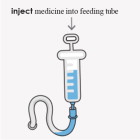
OPTION 2 To administer by feeding tube:
Important:
- Do not use any foods or liquids other than sterile water to mix FIRDAPSE.
- Use only an oral syringe with a catheter tip to give FIRDAPSE through the feeding tube. Talk to your healthcare provider about the size catheter tipped syringe you should use.
How should I store FIRDAPSE?
Firdapse tablets:
- Store FIRDAPSE tablets at room temperature between 68ºF to 77ºF (20ºC to 25ºC).
- Safely throw away FIRDAPSE tablets that are out of date or no longer needed.
Firdapse prepared suspension:
- Store FIRDAPSE prepared oral suspension in the refrigerator between 36°F to 46°F (2°C to 8°C) between doses for up to 24 hours.
- Safely throw away unused FIRDAPSE oral suspension after 24 hours.
Distributed by Catalyst Pharmaceuticals, Inc., Coral Gables, FL 33134
For more information, go to www.YourCatalystPathways.com or call 1-833-422-8259
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Approved: 5/2024
- PRINCIPAL DISPLAY PANEL - 10 mg Tablet Blister Pack Carton
-
INGREDIENTS AND APPEARANCE
FIRDAPSE
amifampridine phosphate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69616-211 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength amifampridine phosphate (UNII: 8HF8FIN815) (amifampridine - UNII:RU4S6E2G0J) amifampridine 10 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) calcium stearate (UNII: 776XM7047L) Product Characteristics Color WHITE (white to off white) Score 2 pieces Shape ROUND Size 10mm Flavor Imprint Code CATALYST;211;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69616-211-06 12 in 1 CARTON 01/07/2019 1 NDC:69616-211-04 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC:69616-211-08 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 01/07/2019 3 NDC:69616-211-03 240 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 01/07/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208078 01/07/2019 Labeler - Catalyst Pharmaceuticals, Inc. (053742248) Establishment Name Address ID/FEI Business Operations Patheon API Manufacturing, Inc. 962538372 API MANUFACTURE(69616-211) , analysis(69616-211) Establishment Name Address ID/FEI Business Operations Catalent Micron Technologies, Inc. 015966157 particle size reduction(69616-211) , ANALYSIS(69616-211) Establishment Name Address ID/FEI Business Operations Pharmaceutics International, Inc. 878265586 MANUFACTURE(69616-211) , analysis(69616-211) Establishment Name Address ID/FEI Business Operations PCI Pharma Services Canada, Inc. 204123822 PACK(69616-211) Establishment Name Address ID/FEI Business Operations Quality Chemical Laboratories, LLC 071344167 ANALYSIS(69616-211) Establishment Name Address ID/FEI Business Operations Patheon Inc. 240769596 ANALYSIS(69616-211) Establishment Name Address ID/FEI Business Operations Patheon Pharmaceuticals Inc. 005286822 MANUFACTURE(69616-211) , PACK(69616-211) Establishment Name Address ID/FEI Business Operations Patheon Softgels Inc. 002193829 ANALYSIS(69616-211) Establishment Name Address ID/FEI Business Operations Robertson-Microlit Laboratories Inc. 177250289 ANALYSIS(69616-211) Establishment Name Address ID/FEI Business Operations SGS Canada Inc. 203668041 analysis(69616-211) Establishment Name Address ID/FEI Business Operations SGS North America Inc. 049859261 analysis(69616-211)