Label: MIFEPRISTONE tablet

- NDC Code(s): 76346-654-03
- Packager: Corcept Therapeutics Incorporated
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 29, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use mifepristone safely and effectively. See full prescribing information for mifepristone.
mifepristone 300 mg tablets
Initial U.S. Approval 2000WARNING: TERMINATION OF PREGNANCY
See full prescribing information for complete boxed warning.
Mifepristone has potent antiprogestational effects and will result in the termination of pregnancy. Pregnancy must therefore be excluded before the initiation of treatment with mifepristone, or if treatment is interrupted for more than 14 days in females of reproductive potential.
RECENT MAJOR CHANGES
Dosage and Administration (2.5) 11/2019 INDICATIONS AND USAGE
Mifepristone is a cortisol receptor blocker indicated to control hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery (1).
- Important Limitations of Use: Do not use for the treatment of type 2 diabetes mellitus unrelated to endogenous Cushing's syndrome.
DOSAGE AND ADMINISTRATION
- Obtain a negative pregnancy test in females of reproductive potential prior to initiating treatment with mifepristone or if treatment is interrupted for more than 14 days. (2.1)
- Administer once daily orally with a meal (2.2).
- The recommended starting dose is 300 mg once daily (2.2).
- Based on clinical response and tolerability, the dose may be increased in 300 mg increments to a maximum of 1200 mg once daily. Do not exceed 20 mg/kg per day (2.2).
- Renal impairment: do not exceed 600 mg once daily (2.3).
- Mild-to-moderate hepatic impairment: do not exceed 600 mg once daily. Do not use in severe hepatic impairment (2.4).
- Concomitant administration with strong CYP3A inhibitors: Do not exceed 900 mg once daily (2.5).
DOSAGE FORMS AND STRENGTHS
Tablets: 300 mg (3)
CONTRAINDICATIONS
- Pregnancy (4, 8.1)
- Patients taking drugs metabolized by CYP3A such as simvastatin, lovastatin, and CYP3A substrates with narrow therapeutic ranges (4)
- Patients receiving systemic corticosteroids for lifesaving purposes (4)
- Women with a history of unexplained vaginal bleeding or endometrial hyperplasia with atypia or endometrial carcinoma (4)
- Patients with known hypersensitivity to mifepristone or to any of the product components (4)
WARNINGS AND PRECAUTIONS
- Adrenal insufficiency: Patients should be closely monitored for signs and symptoms of adrenal insufficiency (5.1).
- Hypokalemia: Hypokalemia should be corrected prior to treatment and monitored for during treatment (5.2).
- Vaginal bleeding and endometrial changes: Women may experience endometrial thickening or unexpected vaginal bleeding. Use with caution if patient also has a hemorrhagic disorder or is on anti-coagulant therapy (5.3).
- QT interval prolongation: Avoid use with QT interval-prolonging drugs, or in patients with potassium channel variants resulting in a long QT interval (5.4).
- Use of Strong CYP3A Inhibitors: Concomitant use can increase mifepristone plasma levels. Use only when necessary and limit mifepristone dose to 900 mg (5.6).
ADVERSE REACTIONS
Most common adverse reactions in Cushing's syndrome (≥ 20%): nausea, fatigue, headache, decreased blood potassium, arthralgia, vomiting, peripheral edema, hypertension, dizziness, decreased appetite, endometrial hypertrophy (6).
To report suspected adverse reactions, contact Corcept Therapeutics at 1-855-844-3270 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Drugs metabolized by CYP3A: Administer drugs that are metabolized by CYP3A at the lowest dose when used with mifepristone (7.1).
- CYP3A inhibitors: Caution should be used when mifepristone is used with strong CYP3A inhibitors. Limit mifepristone dose to 900 mg per day when used with strong CYP3A inhibitors (7.2).
- CYP3A inducers: Do not use mifepristone with CYP3A inducers (7.3).
- Drugs metabolized by CYP2C8/2C9: Use the lowest dose of CYP2C8/2C9 substrates when used with mifepristone (7.4).
- Drugs metabolized by CYP2B6: Use of mifepristone should be done with caution with bupropion and efavirenz (7.5).
- Hormonal contraceptives: Do not use with mifepristone (7.6).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: TERMINATION OF PREGNANCY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to and During mifepristone Administration
2.2 Adult Dosage
2.3 Dosing in Renal Impairment
2.4 Dosing in Hepatic Impairment
2.5 Concomitant Administration with CYP3A Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Adrenal Insufficiency
5.2 Hypokalemia
5.3 Vaginal Bleeding and Endometrial Changes
5.4 QT Interval Prolongation
5.5 Exacerbation/Deterioration of Conditions Treated with Corticosteroids
5.6 Use of Strong CYP3A Inhibitors
5.7 Pneumocystis jiroveci Infection
5.8 Potential Effects of Hypercortisolemia
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs Metabolized by CYP3A
7.2 CYP3A Inhibitors
7.3 CYP3A Inducers
7.4 Drugs Metabolized by CYP2C8/2C9
7.5 Drugs Metabolized by CYP2B6
7.6 Use of Hormonal Contraceptives
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Cushing's Syndrome
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Importance of Preventing Pregnancy
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: TERMINATION OF PREGNANCY
Mifepristone is a potent antagonist of progesterone and cortisol via the progesterone and glucocorticoid (GR-II) receptors, respectively. The antiprogestational effects will result in the termination of pregnancy. Pregnancy must therefore be excluded before the initiation of treatment with mifepristone and prevented during treatment and for one month after stopping treatment by the use of a non-hormonal medically acceptable method of contraception unless the patient has had a surgical sterilization, in which case no additional contraception is needed. Pregnancy must also be excluded if treatment is interrupted for more than 14 days in females of reproductive potential.
-
1 INDICATIONS AND USAGE
Mifepristone is a cortisol receptor blocker indicated to control hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery.
LIMITATIONS OF USE:
- Mifepristone should not be used in the treatment of patients with type 2 diabetes unless it is secondary to Cushing's syndrome.
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to and During mifepristone Administration
Obtain a negative pregnancy test in females of reproductive potential prior to initiating treatment with mifepristone or if treatment is interrupted for more than 14 days [see Contraindications (4), Warnings and Precautions (5.2), Use in Specific Populations (8.1, 8.3)].
2.2 Adult Dosage
The recommended starting dose is 300 mg orally once daily. Mifepristone must be given as a single daily dose. Mifepristone should always be taken with a meal. Patients should swallow the tablet whole. Do not split, crush, or chew tablets.
Dosing and titration
The daily dose of mifepristone may be increased in 300 mg increments. The dose of mifepristone may be increased to a maximum of 1200 mg once daily but should not exceed 20 mg/kg per day. Increases in dose should not occur more frequently than once every 2-4 weeks. Decisions about dose increases should be based on a clinical assessment of tolerability and degree of improvement in Cushing's syndrome manifestations. Changes in glucose control, anti-diabetic medication requirements, insulin levels, and psychiatric symptoms may provide an early assessment of response (within 6 weeks) and may help guide early dose titration. Improvements in cushingoid appearance, acne, hirsutism, striae, and body weight occur over a longer period of time and, along with measures of glucose control, may be used to determine dose changes beyond the first 2 months of therapy. Careful and gradual titration of mifepristone accompanied by monitoring for recognized adverse reactions [See Warnings and Precautions (5.1) and (5.2)] may reduce the risk of severe adverse reactions. Dose reduction or even dose discontinuation may be needed in some clinical situations. If mifepristone treatment is interrupted, it should be reinitiated at the lowest dose (300 mg). If treatment was interrupted because of adverse reactions, the titration should aim for a dose lower than the one that resulted in treatment interruption.
2.3 Dosing in Renal Impairment
No change in initial dose of mifepristone is required in renal impairment. The maximum dose should be limited to 600 mg. [See Renal Impairment (8.6) and Clinical Pharmacology (12.3)]
2.4 Dosing in Hepatic Impairment
No change in the initial dose of mifepristone is required in mild to moderate hepatic impairment. The maximum dose should be limited to 600 mg. Mifepristone should not be used in severe hepatic impairment. [See Hepatic Impairment (8.7) and Clinical Pharmacology (12.3)]
2.5 Concomitant Administration with CYP3A Inhibitors
Ketoconazole and other strong inhibitors of CYP3A, such as itraconazole, nefazodone, ritonavir, nelfinavir, indinavir, atazanavir, amprenavir and fosamprenavir, clarithromycin, conivaptan, lopinavir/ritonavir, posaconazole, saquinavir, telithromycin, or voriconazole may increase exposure to mifepristone. Mifepristone should be used in combination with strong CYP3A inhibitors only when necessary. [See Warnings and Precautions (5.6), Drug Interactions (7.2)]
Administration of mifepristone to patients already being treated with strong CYP3A inhibitors:
- Start at a dose of 300 mg. If clinically indicated, titrate to a maximum of 900 mg.
Administration of strong CYP3A inhibitors to patients already being treated with mifepristone:
- Adjust the dose of mifepristone according to Table 1.
Table 1. Dose adjustment of mifepristone when strong CYP3A inhibitor is added Current dose of mifepristone Adjustment to dose of mifepristone
if adding a strong CYP3A inhibitor300 mg No change 600 mg Reduce dose to 300 mg. If clinically indicated,
titrate to a maximum of 600 mg900 mg Reduce dose to 600 mg. If clinically indicated,
titrate to a maximum of 900 mg1200 mg Reduce dose to 900 mg - 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Mifepristone is contraindicated in:
- Pregnancy [See Dosage and Administration (2.1), Use in Specific Populations (8.1,8.3)]
- Patients taking drugs metabolized by CYP3A such as simvastatin, lovastatin, and CYP3A substrates with narrow therapeutic ranges, such as cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus, due to an increased risk of adverse events. [See Drug Interactions (7.1) and Clinical Pharmacology (12.3)]
- Patients receiving systemic corticosteroids for lifesaving purposes (e.g., immunosuppression after organ transplantation) because mifepristone antagonizes the effect of glucocorticoids.
- Women with a history of unexplained vaginal bleeding or with endometrial hyperplasia with atypia or endometrial carcinoma.
- Patients with known hypersensitivity to mifepristone or to any of the product components.
-
5 WARNINGS AND PRECAUTIONS
5.1 Adrenal Insufficiency
Patients receiving mifepristone may experience adrenal insufficiency. Because serum cortisol levels remain elevated and may even increase during treatment with mifepristone, serum cortisol levels do not provide an accurate assessment of hypoadrenalism in patients receiving mifepristone. Patients should be closely monitored for signs and symptoms of adrenal insufficiency, including weakness, nausea, increased fatigue, hypotension, and hypoglycemia. If adrenal insufficiency is suspected, discontinue treatment with mifepristone immediately and administer glucocorticoids without delay. High doses of supplemental glucocorticoids may be needed to overcome the glucocorticoid receptor blockade produced by mifepristone. Factors considered in deciding on the duration of glucocorticoid treatment should include the long half-life of mifepristone (85 hours).
Treatment with mifepristone at a lower dose can be resumed after resolution of adrenal insufficiency. Patients should also be evaluated for precipitating causes of hypoadrenalism (infection, trauma, etc.).
5.2 Hypokalemia
In a study of patients with Cushing's syndrome, hypokalemia was observed in 44% of subjects during treatment with mifepristone. Hypokalemia should be corrected prior to initiating mifepristone. During mifepristone administration, serum potassium should be measured 1 to 2 weeks after starting or increasing the dose of mifepristone and periodically thereafter. Hypokalemia can occur at any time during mifepristone treatment. Mifepristone-induced hypokalemia should be treated with intravenous or oral potassium supplementation based on event severity. If hypokalemia persists in spite of potassium supplementation, consider adding mineralocorticoid antagonists.
5.3 Vaginal Bleeding and Endometrial Changes
Being an antagonist of the progesterone receptor, mifepristone promotes unopposed endometrial proliferation that may result in endometrium thickening, cystic dilatation of endometrial glands, and vaginal bleeding. Mifepristone should be used with caution in women who have hemorrhagic disorders or are receiving concurrent anticoagulant therapy. Women who experience vaginal bleeding during mifepristone treatment should be referred to a gynecologist for further evaluation.
5.4 QT Interval Prolongation
Mifepristone and its metabolites block IKr. Mifepristone prolongs the QTc interval in a dose-related manner. There is little or no experience with high exposure, concomitant dosing with other QT-prolonging drugs, or potassium channel variants resulting in a long QT interval. [See Warnings & Precautions (5.6)] To minimize risk, the lowest effective dose should always be used.
5.5 Exacerbation/Deterioration of Conditions Treated with Corticosteroids
Use of mifepristone in patients who receive corticosteroids for other conditions (e.g., autoimmune disorders) may lead to exacerbation or deterioration of such conditions, as mifepristone antagonizes the desired effects of glucocorticoid in these clinical settings. For medical conditions in which chronic corticosteroid therapy is lifesaving (e.g., immunosuppression in organ transplantation), mifepristone is contraindicated. [See Contraindications (4.3)]
5.6 Use of Strong CYP3A Inhibitors
Mifepristone should be used with caution in patients taking ketoconazole and other strong inhibitors of CYP3A, such as itraconazole, nefazodone, ritonavir, nelfinavir, indinavir, atazanavir, amprenavir, fosamprenavir, clarithromycin, conivaptan, lopinavir/ritonavir, posaconazole, saquinavir, telithromycin, or voriconazole, as these could increase the concentration of mifepristone in the blood. The benefit of concomitant use of these agents should be carefully weighed against the potential risks. Mifepristone should be used in combination with strong CYP3A inhibitors only when necessary, and in such cases the dose should be limited to 900 mg per day. [See Warnings & Precautions (5.4), Drug Interactions (7.2), and Clinical Pharmacology (12.3)]
5.7 Pneumocystis jiroveci Infection
Patients with endogenous Cushing's syndrome are at risk for opportunistic infections such as Pneumocystis jiroveci pneumonia during mifepristone treatment. Patients may present with respiratory distress shortly after initiation of mifepristone. Appropriate diagnostic tests should be undertaken and treatment for Pneumocystis jiroveci should be considered.
5.8 Potential Effects of Hypercortisolemia
Mifepristone does not reduce serum cortisol levels. Elevated cortisol levels may activate mineralocorticoid receptors which are also expressed in cardiac tissues. Caution should be used in patients with underlying heart conditions including heart failure and coronary vascular disease.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
Safety data on the use of mifepristone are available from 50 patients with Cushing's syndrome enrolled in an uncontrolled, open-label, multi-center trial (Study 400). Forty-three patients had Cushing's disease and all except one had previously undergone pituitary surgery. Four patients had ectopic ACTH secretion, and three had adrenal carcinoma. Patients were treated for up to 24 weeks. A dose of 300 mg per day was administered for the initial 14 days; thereafter, the dose could be escalated in increments of 300 mg per day based on assessments of tolerability and clinical response. Doses were escalated up to 900 mg per day for patients <60 kg, or 1200 mg per day for patients >60 kg.
The most frequently reported adverse reactions (reported in ≥20% of patients, regardless of relationship to mifepristone) were nausea, fatigue, headache, decreased blood potassium, arthralgia, vomiting, peripheral edema, hypertension, dizziness, decreased appetite, and endometrial hypertrophy. Drug-related adverse events resulted in dose interruption or reduction in study drug in 40% of patients.
The adverse reactions that occurred in ≥10% of the Cushing's syndrome patients receiving mifepristone, regardless of relationship to mifepristone, are shown in Table 2.
Table 2. Treatment Emergent Adverse Events Occurring in ≥10% of Cushing's Syndrome Patients Receiving mifepristone Body System/Adverse Reaction Percent (%) of Patients Reporting Event
(n = 50)*The denominator was 26 females who had baseline and end-of-trial transvaginal ultrasound
Gastrointestinal disorders Nausea 48 Vomiting 26 Dry mouth 18 Diarrhea 12 Constipation 10 General disorders and administration/site conditions Fatigue 48 Edema peripheral 26 Pain 14 Nervous system disorders Headache 44 Dizziness 22 Somnolence 10 Musculoskeletal and connective tissue disorders Arthralgia 30 Back pain 16 Myalgia 14 Pain in extremity 12 Investigations Blood potassium decreased 34 Thyroid function test abnormal 18 Infections and infestations Sinusitis 14 Nasopharyngitis 12 Metabolism and nutrition disorders Decreased appetite 20 Anorexia 10 Vascular disorders Hypertension 24 Reproductive system and breast disorders Endometrial hypertrophy 38* Respiratory, thoracic, and mediastinal disorders Dyspnea 16 Psychiatric disorders Anxiety 10 Laboratory Tests
Reductions in high density lipoprotein-cholesterol (HDL-C) levels have been observed following treatment with mifepristone. In study subjects that experienced declines in HDL-C, levels returned to baseline following discontinuation of drug. The clinical significance of the treatment-related reduction in HDL-C levels in patients with Cushing's syndrome is not known.
In a study of patients with Cushing's syndrome, hypokalemia was observed in 44% of subjects during treatment with mifepristone. In these cases, hypokalemia responded to treatment with potassium supplementation and/or mineralocorticoid antagonist therapy (e.g., spironolactone or eplerenone). Hypokalemia should be corrected prior to initiating mifepristone. [See Warnings and Precautions (5.2)]
Elevations of thyroid-stimulating hormone (TSH) were seen in subjects treated with mifepristone. Of the 42 subjects with detectable TSH at baseline, eight (19%) had increases in TSH above the normal range, while remaining asymptomatic. The TSH levels returned to normal in most patients without intervention when mifepristone was discontinued at the end of the study.
Vaginal Bleeding and Endometrial Changes
In Study 400, the thickness of the endometrium increased from a mean of 6.14 mm at baseline (n=23) to 15.7 mm at end-of-trial (n=18) in premenopausal women; in postmenopausal women the increase was from 2.75 mm (n=6) to 7.35 mm (n=8). Endometrial thickness above the upper limit of normal was reported in 10/26 females who had baseline and end-of-trial transvaginal ultrasound (38%). The endometrial thickness returned to the normal range in 3 out of 10 patients 6 weeks after treatment cessation at the end of the study. Vaginal bleeding occurred in 5 out of 35 females (14%). Two of five subjects with vaginal bleeding had normal endometrial thickness. Endometrial biopsies were performed in six patients; five of these patients had endometrial thickening. No endometrial carcinoma was detected in the sampled cases.
Additional Data from Clinical Trials
The following are adverse events that were reported in Study 400 at frequencies of ≥ 5% to 10%, and may be related to mifepristone's mechanism of action:
Gastrointestinal disorders: gastroesophageal reflux, abdominal pain
General disorders and administration site conditions: asthenia, malaise, edema, pitting edema, thirst
Investigations: blood triglycerides increased
Metabolism and nutrition disorders: hypoglycemia
Musculoskeletal and connective tissue disorders: muscular weakness, flank pain, musculoskeletal chest pain
Psychiatric disorders: insomnia
Reproductive system and breast disorders: vaginal hemorrhage, metrorrhagia [See Warnings and Precautions (5.3)]
Adrenal Insufficiency
Adrenal insufficiency was reported in two subjects (4%) in Study 400. The most typical symptoms of adrenal insufficiency were nausea and decreased appetite. No hypotension or hypoglycemia was reported during the events. Adrenal insufficiency resolved in both cases with mifepristone interruption and /or dexamethasone administration.
6.2 Postmarketing Experience
The following adverse reaction has been identified during post approval use of mifepristone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- -
- Angioedema
-
7 DRUG INTERACTIONS
Based on the long terminal half-life of mifepristone after reaching steady state, at least 2 weeks should elapse after cessation of mifepristone before initiating or increasing the dose of any interacting concomitant medication.
7.1 Drugs Metabolized by CYP3A
Because mifepristone is an inhibitor of CYP3A, concurrent use of mifepristone with a drug whose metabolism is largely or solely mediated by CYP3A is likely to result in increased plasma concentrations of the drug. Discontinuation or dose reduction of such medications may be necessary with mifepristone co-administration.
Mifepristone increased the exposure to simvastatin and simvastatin acid significantly in healthy subjects. Concomitant use of simvastatin or lovastatin is contraindicated because of the increased risk of myopathy and rhabdomyolysis. [See Contraindications (4.2), Clinical Pharmacology 12.3]
The exposure of other substrates of CYP3A with narrow therapeutic ranges, such as cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus, may be increased by concomitant administration with mifepristone. Therefore, the concomitant use of such CYP3A substrates with mifepristone is contraindicated. [See Contraindications (4.2)]
Other drugs with similar high first pass metabolism in which CYP3A is the primary route of metabolism should be used with extreme caution if co-administered with mifepristone. The lowest possible dose and/or a decreased frequency of dosing must be used with therapeutic drug monitoring when possible. Use of alternative drugs without these metabolic characteristics is advised when possible with concomitant mifepristone.
If drugs that undergo low first pass metabolism by CYP3A or drugs in which CYP3A is not the major metabolic route are co-administered with mifepristone, use the lowest dose of concomitant medication necessary, with appropriate monitoring and follow-up. [See Clinical Pharmacology (12.3)]
7.2 CYP3A Inhibitors
Medications that inhibit CYP3A could increase plasma mifepristone concentrations and dose reduction of mifepristone may be required.
Ketoconazole and other strong inhibitors of CYP3A, such as itraconazole, nefazodone, ritonavir, nelfinavir, indinavir, atazanavir, amprenavir and fosamprenavir, clarithromycin, conivaptan, lopinavir /ritonavir, posaconazole, saquinavir, telithromycin, or voriconazole may increase exposure to mifepristone. Caution should be used when strong CYP3A inhibitors are prescribed in combination with mifepristone. The benefit of concomitant use of these agents should be carefully weighed against the potential risks. The dose of mifepristone should be limited to 900 mg, and strong inhibitors of CYP3A should be used only when necessary. [See Dosage and Administration (2.4), Warnings & Precautions (5.6), and Clinical Pharmacology (12.3)]
7.3 CYP3A Inducers
No medications that induce CYP3A have been studied when co-administered with mifepristone. Avoid co-administration of mifepristone and CYP3A inducers such as rifampin, rifabutin, rifapentin, phenobarbital, phenytoin, carbamazepine, and St. John's wort.
7.4 Drugs Metabolized by CYP2C8/2C9
Because mifepristone is an inhibitor of CYP2C8/2C9, concurrent use of mifepristone with a drug whose metabolism is largely or solely mediated by CYP2C8/2C9 is likely to result in increased plasma concentrations of the drug.
Mifepristone significantly increased exposure of fluvastatin, a typical CYP2C8/2C9 substrate, in healthy subjects. When given concomitantly with mifepristone, drugs that are substrates of CYP2C8/2C9 (including non-steroidal anti-inflammatory drugs, warfarin, and repaglinide) should be used at the smallest recommended doses, and patients should be closely monitored for adverse effects. [See Clinical Pharmacology (12.3)]
7.5 Drugs Metabolized by CYP2B6
Mifepristone is an inhibitor of CYP2B6 and may cause significant increases in exposure of drugs that are metabolized by CYP2B6 such as bupropion and efavirenz. Since no study has been conducted to evaluate the effect of mifepristone on substrates of CYP2B6, the concomitant use of bupropion and efavirenz should be undertaken with caution. [See Clinical Pharmacology (12.3)]
7.6 Use of Hormonal Contraceptives
Mifepristone is a progesterone-receptor antagonist and will interfere with the effectiveness of hormonal contraceptives. Therefore, non-hormonal contraceptive methods should be used. [See Use In Specific Populations (8.3)]
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Mifepristone is contraindicated in pregnancy because the use of mifepristone results in pregnancy loss. There are no data that assess the risk of birth defects in women exposed to mifepristone during pregnancy. Available data limited to exposure following a single dose of mifepristone during pregnancy showed a higher rate of major birth defects compared to the general population comparator (See Data). Mifepristone administered to pregnant mice, rats, and rabbits during organogenesis caused pregnancy loss in all species at clinically relevant doses based on body surface area comparisons (See Data). The inhibition of both endogenous and exogenous progesterone by mifepristone at the progesterone receptor results in pregnancy loss. If mifepristone is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus. [See Contraindications (4)]
The estimated risk of fetal loss is elevated in patients with active Cushing's syndrome (24-30%), and the risk of major birth defects is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Data
Human Data
There are no data on long term exposure to mifepristone in pregnancy. Available data are limited to exposure to a single dose of mifepristone for pregnancy termination. In a prospective study in France of 46 pregnancies exposed to a single dose of mifepristone alone and 59 pregnancies exposed to a single dose of mifepristone and misoprostol, the overall major birth defect rate (4%) was greater than the general population background rate of 2 to 3% (2 birth defects in each group). There was no pattern of birth defects identified.
Animal Data
Reproductive studies were performed in mice, rats and rabbits at doses of 0.25 to 4.0 mg/kg (less than human exposure at the maximum clinical dose, based on body surface area). Because of the anti-progestational activity of mifepristone, fetal losses were much higher than in control animals. Skull deformities were detected in rabbit studies at less than human exposure, although mifepristone did not cause any adverse developmental effects in rats or mice during organogenesis. These deformities were most likely due to the mechanical effects of uterine contractions resulting from antagonism of the progesterone receptor.
8.2 Lactation
Risk Summary
Mifepristone is present in human milk, however, there are no data on the amount of mifepristone in human milk, the effects on the breastfed infant, or the effects on milk production during long term use of mifepristone (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for mifepristone and any potential adverse effects on the breastfed child from mifepristone or from the underlying maternal condition.
Clinical Considerations
To minimize exposure to a breastfed infant, women who discontinue or interrupt mifepristone treatment may consider pumping and discarding milk during treatment and for 18-21 days (5-6 half-lives) after the last dose, before breastfeeding.
Data
Available published data based on intake of a single dose of 600 mg of mifepristone in 10 breastfeeding women who were 6-12 months postpartum showed a small amount in breast milk (the estimated relative infant dose was 0.5%). The half-life of mifepristone is longer with repeat dosing compared to a single dose; therefore, there may be greater exposure with long term use.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Due to its anti-progestational activity, mifepristone causes pregnancy loss. Perform pregnancy testing before the initiation of treatment with mifepristone or if treatment is interrupted for more than 14 days in females of reproductive potential.
Contraception
Recommend non-hormonal contraception for the duration of treatment and for one month after stopping treatment. Mifepristone interferes with the effectiveness of hormonal contraceptives. [See Drug Interactions (7.6)]
8.4 Pediatric Use
Safety and effectiveness of mifepristone in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies with mifepristone did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger people.
8.6 Renal Impairment
The maximum dose should not exceed 600 mg per day in renally impaired patients. [See Clinical Pharmacology (12.3)]
8.7 Hepatic Impairment
In patients with mild to moderate hepatic impairment, the maximum dose should not exceed 600 mg per day. The pharmacokinetics of mifepristone in patients with severe hepatic impairment has not been studied, and mifepristone should not be used in these patients. [See Clinical Pharmacology (12.3)]
- 10 OVERDOSAGE
-
11 DESCRIPTION
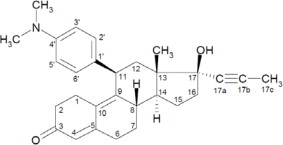
Mifepristone is a cortisol receptor blocker for oral administration. The chemical name of mifepristone is 11β-(4-dimethylaminophenyl)-17β-hydroxy-17α-(1-propynyl)-estra-4, 9-dien-3-one. The chemical formula is C29H35NO2; the molecular weight is 429.60, and the structural formula is:
Mifepristone demonstrates a pH-related solubility profile. The greatest solubility is achieved in acidic media (~ 25 mg/mL at pH 1.5) and solubility declines rapidly as the pH is increased. At pH values above 2.5 the solubility of mifepristone is less than 1 mg/mL.
Each mifepristone tablet for oral use contains 300 mg of mifepristone. The inactive ingredients of mifepristone tablets are silicified microcrystalline cellulose, sodium starch glycolate, hydroxypropylcellulose, sodium lauryl sulfate, magnesium stearate, hypromellose, titanium dioxide, triacetin, D&C yellow 10 aluminum lake, polysorbate 80, and FD&C yellow 6 aluminum lake.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mifepristone is a selective antagonist of the progesterone receptor at low doses and blocks the glucocorticoid receptor (GR-II) at higher doses. Mifepristone has high affinity for the GR-II receptor but little affinity for the GR-I (MR, mineralocorticoid) receptor. In addition, mifepristone appears to have little or no affinity for estrogen, muscarinic, histaminic, or monoamine receptors.
12.2 Pharmacodynamics
Because mifepristone acts at the receptor level to block the effects of cortisol, its antagonistic actions affect the hypothalamic-pituitary-adrenal (HPA) axis in such a way as to further increase circulating cortisol levels while, at the same time, blocking their effects.
Mifepristone and the three active metabolites have greater affinity for the glucocorticoid receptor [100% (mifepristone), 61% (metabolite 1), 48% (metabolite 2), and 45% (metabolite 3)] than either dexamethasone (23%) or cortisol (9%).
12.3 Pharmacokinetics
Absorption
Following oral administration, time to peak plasma concentrations of mifepristone occurred between 1 and 2 hours following single dose, and between 1 and 4 hours following multiple doses of 600 mg of mifepristone in healthy volunteers. Mean plasma concentrations of three active metabolites of mifepristone peak between 2 and 8 hours after multiple doses of 600 mg/day, and the combined concentrations of the metabolites exceed that of the parent mifepristone. Exposure to mifepristone is substantially less than dose proportional. Time to steady state is within 2 weeks, and the mean (SD) half-life of the parent mifepristone was 85 (61) hours following multiple doses of 600 mg/day of mifepristone.
Studies evaluating the effects of food on the pharmacokinetics of mifepristone demonstrate a significant increase in plasma levels of mifepristone when dosed with food. To achieve consistent plasma drug concentrations, patients should be instructed to always take their medication with meals.
Distribution
Mifepristone is highly bound to alpha-1-acid glycoprotein (AAG) and approaches saturation at doses of 100 mg (2.5 μM) or more. Mifepristone and its metabolites also bind to albumin and are distributed to other tissues, including the central nervous system (CNS). As determined in vitro by equilibrium dialysis, binding of mifepristone and its three active metabolites to human plasma proteins was concentration-dependent. Binding was approximately 99.2% for mifepristone, and ranged from 96.1 to 98.9% for the three active metabolites at clinically relevant concentrations.
Metabolism
Cytochrome P450 3A4 (CYP3A4) has been shown to be involved in mifepristone metabolism in human liver microsomes. Two of the known active metabolites are the product of demethylation (one monodemethylated and one di-demethylated), while a third active metabolite results from hydroxylation (monohydroxylated).
Renal Impairment
The pharmacokinetics of mifepristone in subjects with severe renal impairment (creatinine clearance [CrCL] < 30 mL/min, but not on dialysis) was evaluated following multiple doses of 1200 mg mifepristone for 7 days. Mean exposure to mifepristone increased 31%, with similar or smaller increases in metabolite exposure as compared to subjects with normal renal function (CrCL ≥ 90 mL/min). There was large variability in the exposure of mifepristone and its metabolites in subjects with severe renal impairment as compared to subjects with normal renal function (geometric least square mean ratio [CI] for AUC of mifepristone: 1.21 [0.71-2.06]; metabolite 1: 1.43 [0.84-2.44]; metabolite 2: 1.18 [0.64-2.17] and metabolite 3: 1.19 [0.71-1.99]). No change in the initial dose of mifepristone is needed for renal impairment; the maximum dose should not exceed 600 mg per day.
Hepatic Impairment
The pharmacokinetics of mifepristone in subjects with moderate hepatic impairment (Child-Pugh Class B) was evaluated in a single- and multiple-dose study (600 mg for 7 days). The pharmacokinetics in subjects with moderate hepatic impairment was similar to those with normal hepatic function. There was large variability in the exposure of mifepristone and its metabolites in subjects with moderate hepatic impairment as compared to subjects with normal hepatic function (geometric least square mean ratio [CI] for AUC of mifepristone: 1.02 [0.59-1.76]; metabolite 1: 0.95 [0.52-1.71]; metabolite 2: 1.37 [0.71-2.62] and metabolite 3: 0.62 [0.33-1.16]). Due to limited information on safety in patients with mild-to-moderate hepatic impairment, the maximum dose should not exceed 600 mg per day. The pharmacokinetics of mifepristone in patients with severe hepatic disease has not been studied. Mifepristone is not recommended in patients with severe hepatic disease.
In Vitro Assessment of Drug Interactions
In vitro studies indicate a potential for CYP-mediated drug interactions by mifepristone and/or its metabolites with substrates of CYP2A6, CYP2C8/2C9, CYP2C19, CYP3A4, CYP1A2, CYP2B6, CYP2D6, and CYP2E1. In vitro studies also indicated an interaction potential for drug transport mediated by P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). In vitro studies indicate mifepristone metabolism is mediated by CYP3A, and that mifepristone also inhibits and induces CYP3A.
In Vivo Assessment of Drug Interactions (see Table 3)
Table 3. Summary Table of mifepristone Drug-Drug Interaction Effects *No effect = 90% CI within range 0.80 – 1.25
†See Section 12.2 for the relative potencies of the three metabolites
1 Simvastatin 40 mg dose used as reference for the comparison. Result could be representative of other oral drugs with CYP3A metabolism and high first pass effect: cyclosporine, midazolam, triazolam, pimozide, sildenafil, sirolimus, and tacrolimus
2 Result could be representative of other oral drugs with CYP3A metabolism and low first pass effect. Clinical significance of any interaction will depend on the therapeutic margin of the drug.
3 Result could be representative of other oral drugs with CYP2C8/C9 metabolism
4 Plasma digoxin concentration should be measured after 1 to 2 weeks of concomitant use and following usual clinical practice at appropriate intervals thereafter.
5Result could be representative of other mild inhibitors of CYP3A
Dosing of
mifepristoneCoadministered
DrugDosing of
Coadministered
DrugGeometric Mean Ratio
(analyte ratio with/without
drug coadministration)
Analyte AUC Cmax Effect of mifepristone on Coadministered Drug Contraindicated with mifepristone [See Contraindications (4)] 1200 mg once daily for
10 dayssimvastatin1 80 mg single dose simvastatin
acid
simvastatin15.70
10.4018.20
7.02Use lowest dose of coadministered drug, based on clinical experience and/or use of therapeutic drug monitoring 1200 mg once daily for
10 daysalprazolam2 1 mg single dose alprazolam
4-hydroxy-alprazolam1.80
0.760.81
0.391200 mg once daily for
7 daysfluvastatin3 40 mg single dose fluvastatin 3.57 1.76 1200 mg once daily for
10 daysdigoxin4 0.125 mg once daily digoxin 1.40 1.64 Effect of Coadministered Drug on mifepristone Dose adjustment required 600 mg once daily for
17 daysketoconazole 200 mg bid on
days 13-17mifepristone
Metabolite 1†
Metabolite 2†
Metabolite 3†1.38
1.02
1.67
0.951.28
1.06
1.69
0.96900 mg once daily for
14 daysitraconazole 200 mg daily for
14 daysmifepristone
Metabolite 1†
Metabolite 2†
Metabolite 3†1.10
1.04
1.23
0.971.20
1.00
1.19
0.94Effect of Coadministered Drug on mifepristone No dosing adjustment required 300 mg once daily for
14 dayscimetidine5 800 mg once daily mifepristone 0.85* 0.75 -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mifepristone was evaluated for carcinogenicity potential in rats and mice. Rats were dosed for up to two years at doses of 5, 25, and 125 mg/kg of mifepristone. The high dose was the maximum tolerated dose, but exposure at all doses was below exposure at the maximum clinical dose based on AUC comparison. Female rats had a statistically significant increase in follicular cell adenomas/carcinomas and liver adenomas. It is plausible that these tumors are due to drug-induced enzyme metabolism, a mechanism not considered clinically relevant, but studies confirming this mechanism were not conducted with mifepristone. Mice were also tested for up to 2 years at mifepristone doses up to the maximum tolerated dose of 125 mg/kg, which provided exposure below the maximum clinical dose based on AUC. No drug-related tumors were seen in mice.
Mifepristone was not genotoxic in a battery of bacterial, yeast, and mammalian in vitro assays, and an in vivo micronucleus study in mice.
The pharmacological activity of mifepristone disrupts the estrus cycle of adult rats at a dose of 0.3 mg/kg (less than human exposure at the maximum clinical dose, based on body surface area). However, following withdrawal of treatment and subsequent resumption of the estrus cycle, there was no effect on reproductive function when mated.
A single subcutaneous dose of mifepristone (up to 100 mg/kg) to rats on the first day after birth did not adversely affect future reproductive function in males or females, although the onset of puberty was slightly premature in dosed females. Repeated doses of mifepristone (1 mg every other day) to neonatal rats resulted in potentially adverse fertility effects, including oviduct and ovary malformations in females, delayed male puberty, deficient male sexual behavior, reduced testicular size, and lowered ejaculation frequency.
-
14 CLINICAL STUDIES
14.1 Cushing's Syndrome
An uncontrolled, open-label, 24-week, multicenter clinical study was conducted to evaluate the safety and efficacy of mifepristone in the treatment of endogenous Cushing's syndrome. The study enrolled 50 subjects with clinical and biochemical evidence of hypercortisolemia despite prior surgical treatment and radiotherapy. The reasons for medical treatment were failed surgery, recurrence of disease, and poor medical candidate for surgery. Forty-three patients (86%) had Cushing's disease, four patients (8%) had ectopic ACTH secretion, and three (6%) had adrenal carcinoma. Baseline characteristics included: mean age of 45 years (range 26 to 71), mean BMI of 36 kg/m2 (range 24 to 66), mean weight 100 kg (range 61 to 199), and mean waist circumference was 119 cm (range 89 to 178); 70% were female; 84% were white and 16% were black or African American. Baseline mean urinary free cortisol level was 365 μg per 24 hr.
Patients belonged to one of two cohorts: a “diabetes” cohort (29 patients, 26 with type 2 diabetes and 3 with glucose intolerance), and a “hypertension” cohort (21 patients). Efficacy was evaluated separately in the two cohorts. Mifepristone treatment was started in all patients at a dose of 300 mg once a day. The study protocol allowed an increase in dose to 600 mg after 2 weeks, and then by additional 300 mg increments every 4 weeks to a maximum of 900 mg per day for patients <60 kg, or 1200 mg per day for patients >60 kg, based on clinical tolerance and clinical response.
Results in the diabetes cohort
Patients in the diabetes cohort underwent standard oral glucose tolerance tests at baseline and periodically during the clinical study. Anti-diabetic medications were allowed but had to be kept stable during the trial and patients had to be on stable anti-diabetic regimens prior to enrollment. The primary efficacy analysis for the diabetes cohort was an analysis of responders. A responder was defined as a patient who had a ≥ 25% reduction from baseline in glucose AUC. The primary efficacy analysis was conducted in the modified intent-to-treat population (n=25) defined as all patients who received a minimum of 30 days on mifepristone. Fifteen of 25 patients (60%) were treatment responders (95% CI: 39%,78%).
Mean HbA1c was 7.4% in the 24 patients with HbA1c values at baseline and Week 24. For these 24 patients mean reduction in HbA1c was 1.1% (95% CI -1.6, -0.7) from baseline to the end of the trial. Fourteen of 24 patients had above normal HbA1c levels at baseline, ranging between 6.7% and 10.4%; all of these patients had reductions in HbA1c by the end of the study (range -0.4 to -4.4%) and eight of 14 patients (57%) normalized HbA1c levels at trial end. Antidiabetic medications were reduced in 7 of the 15 DM subjects taking antidiabetic medication and remained constant in the others.
Results in the hypertension cohort
There were no changes in mean systolic and diastolic blood pressures at the end of the trial relative to baseline in the modified intent-to-treat population (n=21).
Signs and symptoms of Cushing's syndrome in both cohorts
Individual patients showed varying degrees of improvement in Cushing's syndrome manifestations such as cushingoid appearance, acne, hirsutism, striae, psychiatric symptoms, and excess total body weight. Because of the variability in clinical presentation and variability of response in this open label trial, it is uncertain whether these changes could be ascribed to the effects of mifepristone.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Mifepristone is supplied as a light yellow to yellow, film-coated, oval-shaped tablet debossed with “Corcept” on one side and “300” on the other. Each tablet contains 300 mg of mifepristone. Mifepristone tablets are available in bottles of 280 tablets (NDC 76346-654-03).
Store at controlled room temperature, 25 °C (77 °F); excursions permitted to 15 to 30 °C (59 – 86 °F). [See USP Controlled Room Temperature]
-
17 PATIENT COUNSELING INFORMATION
As a part of patient counseling, doctors must review the mifepristone Medication Guide with every patient.
17.1 Importance of Preventing Pregnancy
- Advise patients that mifepristone will cause termination of pregnancy. Mifepristone is contraindicated in pregnant patients.
- Mifepristone reduces the effectiveness of hormonal contraceptives. Counsel females of reproductive potential regarding pregnancy prevention and planning with a non-hormonal contraceptive prior to use of mifepristone and up to one month after the end of treatment.
- Instruct patients to contact their physician immediately if they suspect or confirm they are pregnant.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration
Revised: 07/2020
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
MIFEPRISTONE
mifepristone tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:76346-654 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MIFEPRISTONE (UNII: 320T6RNW1F) (MIFEPRISTONE - UNII:320T6RNW1F) MIFEPRISTONE 300 mg Inactive Ingredients Ingredient Name Strength SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TRIACETIN (UNII: XHX3C3X673) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) ALUMINUM OXIDE (UNII: LMI26O6933) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) Product Characteristics Color yellow (YELLOW) Score no score Shape OVAL (OVAL) Size 16mm Flavor Imprint Code CORCEPT;300 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:76346-654-03 280 in 1 BOTTLE; Type 0: Not a Combination Product 05/28/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202107 05/28/2024 Labeler - Corcept Therapeutics Incorporated (069540610)