Label: NATEGLINIDE tablet




- NDC Code(s): 60687-673-11, 60687-673-21, 60687-684-11, 60687-684-21
- Packager: American Health Packaging
- This is a repackaged label.
- Source NDC Code(s): 64380-167, 64380-168
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 28, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NATEGLINIDE TABLETS safely and effectively. See full prescribing information for NATEGLINIDE TABLETS.
NATEGLINIDE tablets, for oral use
Initial U.S. Approval: 2000INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- Recommended dose is 120 mg three times daily (2)
- In patients who are near glycemic goal when treatment is initiated, 60 mg three times daily may be administered. (2)
- Administer 1 to 30 minutes before meals (2)
- If a meal is skipped, skip the scheduled dose to reduce the risk of hypoglycemia. (2, 5.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 60 mg and 120 mg (3)
CONTRAINDICATIONS
- History of hypersensitivity to nateglinide or its inactive ingredients (4)
WARNINGS AND PRECAUTIONS
- Hypoglycemia: Nateglinide may cause hypoglycemia. Administer before meals to reduce the risk of hypoglycemia. Skip the scheduled dose of nateglinide if a meal is skipped to reduce the risk of hypoglycemia. (5.1)
- Macrovascular Outcomes: There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with nateglinide. (5.2)
ADVERSE REACTIONS
- Common adverse reactions associated with nateglinide (3% or greater incidence) were upper respiratory tract infection, back pain, flu symptoms, dizziness, arthropathy, diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Strides Pharma Inc at 1-877-244-9825 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Drugs That May Increase the Potential for Hypoglycemia: Nateglinide dose reductions and increased frequency of glucose monitoring may be required when co-administered (7)
- Drugs That May Increase the Potential for Hyperglycemia: Nateglinide dose increases and increased frequency of glucose monitoring may be required when co-administered (7)
- Drugs That May Blunt Signs and Symptoms of Hypoglycemia: Increased frequency of glucose monitoring may be required when co-administered (7)
USE IN SPECIFIC POPULATIONS
- Lactation: Nateglinide is not recommended when breastfeeding (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypoglycemia
5.2 Macrovascular Outcomes
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Monotherapy
14.2 Monotherapy Compared to Glyburide
14.3 Monotherapy and In Combination with Metformin
14.4 Add-On Combination Therapy with Rosiglitazone
14.5 Add-On Combination Therapy with Glyburide
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
The recommended dose of nateglinide is 120 mg orally three times daily before meals.
The recommended dose of nateglinide is 60 mg orally three times daily before meals in patients who are near glycemic goal when treatment is initiated.
Instruct patients to take nateglinide 1 to 30 minutes before meals.
In patients who skip meals, instruct patients to skip the scheduled dose of nateglinide to reduce the risk of hypoglycemia [see Warnings and Precautions (5.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypoglycemia
All glinides, including nateglinide, can cause hypoglycemia [see Adverse Reactions (6.1)]. Severe hypoglycemia can cause seizures, may be life-threatening, or cause death. Hypoglycemia can impair concentration ability and reaction time; this may place an individual and others at risk in situations where these abilities are important (e.g., driving or operating other machinery).
Hypoglycemia can happen suddenly and symptoms may differ in each individual and change over time in the same individual. Symptomatic awareness of hypoglycemia may be less pronounced in patients with longstanding diabetes, in patients with diabetic neuropathy (nerve disease), in patients using medications that block the sympathetic nervous system (e.g., beta-blockers) [see Drug Interactions (7)], or in patients who experience recurrent hypoglycemia.
Factors which may increase the risk of hypoglycemia include changes in meal pattern (e.g., macronutrient content), changes in level of physical activity, changes to coadministered medication [see Drug Interactions (7)], and concomitant use with other antidiabetic agents. Patients with renal or hepatic impairment may be at higher risk of hypoglycemia [see Use in Specific Populations (8.6, 8.7), Clinical Pharmacology (12.3)].
Patients should take nateglinide before meals and be instructed to skip the dose of nateglinide if a meal is skipped [see Dosage and Administration (2)]. Patients and caregivers must be educated to recognize and manage hypoglycemia. Self-monitoring of blood glucose plays an essential role in the prevention and management of hypoglycemia. In patients at higher risk for hypoglycemia and patients who have reduced symptomatic awareness of hypoglycemia, increased frequency of blood glucose monitoring is recommended.
-
6 ADVERSE REACTIONS
The following serious adverse reaction is also described elsewhere in the labeling:
- Hypoglycemia [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials, approximately 2,600 patients with type 2 diabetes mellitus were treated with nateglinide. Of these, approximately 1,335 patients were treated for 6 months or longer and approximately 190 patients for one year or longer. Table 1 shows the most common adverse reactions associated with nateglinide.
Table 1: Adverse Reactions other than Hypoglycemia (%) occurring Greater than or Equal to 2% in Nateglinide-Treated Patients from Pool of 12 to 64 week Placebo Controlled Trials Placebo
N = 458
Nateglinide
N = 1441
Preferred Term
Upper Respiratory Infection
8.1
10.5
Back Pain
3.7
4.0
Flu Symptoms
2.6
3.6
Dizziness
2.2
3.6
Arthropathy
2.2
3.3
Diarrhea
3.1
3.2
Accidental Trauma
1.7
2.9
Bronchitis
2.6
2.7
Coughing
2.2
2.4
Hypoglycemia
Episodes of severe hypoglycemia (plasma glucose less than 36 mg/dL) were reported in two patients treated with nateglinide. Non-severe hypoglycemia occurred in 2.4 % of nateglinide treated patients and 0.4 % of placebo-treated patients [see Warnings and Precautions (5.1)].Weight Gain
Patients treated with nateglinide had statistically significant mean increases in weight compared to placebo. In clinical trials, the mean weight increases with nateglinide 60 mg (3 times daily) and nateglinide 120 mg (3 times daily) compared to placebo were 1.0 kg and 1.6 kg, respectively.Laboratory Test
Increases in Uric Acid: There were increases in mean uric acid levels for patients treated with nateglinide alone, nateglinide in combination with metformin, metformin alone, and glyburide alone. The respective differences from placebo were 0.29 mg/dL, 0.45 mg/dL, 0.28 mg/dL, and 0.19 mg/dL.6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of nateglinide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity Reactions: Rash, itching, and urticaria
- Hepatobiliary Disorders: Jaundice, cholestatic hepatitis, and elevated liver enzymes
-
7 DRUG INTERACTIONS
Table 2 includes a list of drugs with clinically important drug interactions when concomitantly administered or withdrawn with nateglinide and instructions for managing or preventing them.
Table 2: Clinically Significant Drug Interactions with Nateglinide Drugs That May Increase the Blood-Glucose-Lowering Effect of Nateglinide and Susceptibility to Hypoglycemia
Drugs:
Nonsteroidal anti-inflammatory drugs (NSAIDs), salicylates, monoamine oxidase inhibitors, non-selective beta-adrenergic-blocking agents, anabolic hormones (e.g., methandrostenolone), guanethidine, gymnema sylvestre, glucomannan, thioctic acid, and inhibitors of CYP2C9 (e.g., amiodarone, fluconazole, voriconazole, sulfinpyrazone) or in patients known to be poor metabolizers of CYP2C9 substrates, alcohol.
Intervention:
Dose reductions and increased frequency of glucose monitoring may be required when nateglinide is coadministered with these drugs.
Drugs and Herbals That May Reduce the Blood-Glucose-Lowering Effect of Nateglinide and Increase Susceptibility to Hyperglycemia
Drugs:
Thiazides, corticosteroids, thyroid products, sympathomimetics, somatropin, somatostatin analogues (e.g., lanreotide, octreotide), and CYP inducers (e.g., rifampin, phenytoin and St John's Wort).
Intervention:
Dose increases and increased frequency of glucose monitoring may be required when nateglinide is coadministered with these drugs.
Drugs That May Blunt Signs and Symptoms of Hypoglycemia
Drugs:
beta-blockers, clonidine, guanethidine, and reserpine
Intervention:
Increased frequency of glucose monitoring may be required when nateglinide is coadministered with these drugs.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data from published literature and the applicant's pharmacovigilance with use of nateglinide in pregnant women are insufficient to identify a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy ( see Clinical Considerations). Nateglinide should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In animal reproduction studies, there was no teratogenicity in rats and rabbits administered oral nateglinide during organogenesis at approximately 27 and 8 times the maximum recommended human dose (MRHD), respectively, based on body surface area (BSA).The estimated background risk of major birth defects is 6% to 10% in women with pre-gestational diabetes with a HbA 1c > 7 and has been reported to be as high as 20% to 25% in women with a HbA 1c > 10. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.Data
Animal data
In embryofetal development studies, nateglinide administered orally during the period of organogenesis was not teratogenic in rats at doses up to 1,000 mg/kg (corresponding to 27 times the MRHD of 120 mg three times per day, based on BSA). In rabbits, embryonic development was adversely affected at 500 mg/kg/day and the incidence of gallbladder agenesis or small gallbladder was increased at a dose of 300 and 500 mg/kg (corresponding to 16 and 27 times the MRHD). No such effects were observed at 150 mg/kg/day (corresponding to 8 times the MRHD). In a pre- and postnatal development study in rats, nateglinide administered by oral gavage at doses of 100, 300, and 1000 mg/kg/day from gestation day 17 to lactation day 21 resulted in lower body weight in offspring of rats administered nateglinide at 1,000 mg/kg/day (corresponding to 27 times the MHRD).8.2 Lactation
Risk summary
There are no data on the presence of nateglinide in human milk, the effects on the breastfeeding infant, or the effects on milk production. The drug is present in animal milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk ( see Data). Because the potential for hypoglycemia in breast-fed infants, advise women that use of nateglinide is not recommended while breastfeeding.Data
In rat reproduction studies, nateglinide and its metabolite are excreted in the milk following oral dose (300 mg/kg). The overall milk: plasma (M/P) concentration ratio of the total radioactivity was approximately 1.4 based on AUC 0-48 values. The M/P ratio of unchanged nateglinide was approximately 2.2.8.4 Pediatric Use
The safety and effectiveness of nateglinide have not been established in pediatric patients.
8.5 Geriatric Use
436 patients 65 years and older, and 80 patients 75 years and older were exposed to nateglinide in clinical studies. No differences were observed in safety or efficacy of nateglinide between patients age 65 and over, and those under age 65. However, greater sensitivity of some older individuals to nateglinide therapy cannot be ruled out.
8.6 Renal Impairment
No dosage adjustment is recommended in patients with mild to severe renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment. Use of nateglinide in patients with moderate-to-severe hepatic impairment has not been studied and therefore, should be used with caution in these patients [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There have been no instances of overdose with nateglinide in clinical trials. However, an overdose may result in an exaggerated glucose-lowering effect with the development of hypoglycemic symptoms. Hypoglycemic symptoms without loss of consciousness or neurological findings should be treated with oral glucose and adjustments in dosage and/or meal patterns. Severe hypoglycemic reactions with coma, seizure, or other neurological symptoms should be treated with intravenous glucose. As nateglinide is highly protein bound, dialysis is not an efficient means of removing it from the blood.
-
11 DESCRIPTION
Nateglinide Tablets, USP are an oral blood glucose-lowering drug of the glinide class. Nateglinide, (-)-N-[(trans-4-isopropylcyclohexane)carbonyl]-D-phenylalanine, is structurally unrelated to the oral sulfonylurea insulin secretagogues.
The structural formula is as shown:
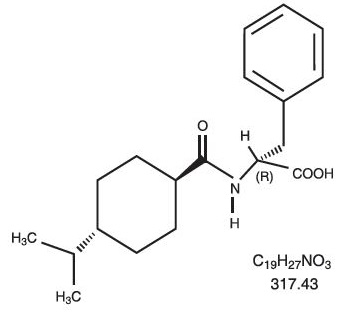
Nateglinide is a white powder with a molecular weight of 317.43. It is freely soluble in methanol, ethanol, and chloroform, soluble in ether, sparingly soluble in acetonitrile and octanol, and practically insoluble in water. Nateglinide biconvex tablets contain 60 mg, or 120 mg, of nateglinide for oral administration.
Inactive Ingredients: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, pregelatinized starch (starch 1500 ®). Starch 1500 ® is partially pregelatinized maize starch. The 60 mg also contains iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. In addition, the 120 mg contains FD&C Yellow #6/Sunset Yellow Aluminum Lake, iron oxide yellow.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nateglinide lowers blood glucose levels by stimulating insulin secretion from the pancreas. This action is dependent upon functioning beta-cells in the pancreatic islets. Nateglinide interacts with the ATP-sensitive potassium (K +ATP) channel on pancreatic beta-cells. The subsequent depolarization of the beta cell opens the calcium channel, producing calcium influx and insulin secretion. The extent of insulin release is glucose dependent and diminishes at low glucose levels. Nateglinide is highly tissue selective with low affinity for heart and skeletal muscle.
12.2 Pharmacodynamics
Nateglinide stimulates pancreatic insulin secretion within 20 minutes of oral administration. When nateglinide is dosed before meals, the peak rise in plasma insulin occurs approximately 1 hour after dosing and falls to baseline by 4 hours after dosing.
12.3 Pharmacokinetics
In patients with Type 2 diabetes, multiple dose administration of nateglinide over the dosage range of 60 mg to 240 mg shows linear pharmacokinetics for both area under the curve (AUC) and C max. In patients with Type 2 diabetes, there is no apparent accumulation of nateglinide upon multiple dosing of up to 240 mg three times daily for 7 days.
Absorption
Absolute bioavailability of nateglinide is approximately 73%. Plasma profiles are characterized by multiple plasma concentration peaks when nateglinide is administered under fasting conditions. This effect is diminished when nateglinide is taken prior to a meal. Following oral administration immediately prior to a meal, the mean peak plasma nateglinide concentrations (C max) generally occur within 1 hour (T max) after dosing. T max is independent of dose.The pharmacokinetics of nateglinide are not affected by the composition of a meal (high protein, fat, or carbohydrate). However, peak plasma levels are significantly reduced when nateglinide is administered 10 minutes prior to a liquid meal as compared to solid meal. When given with or after meals, the extent of nateglinide absorption (AUC) remains unaffected. However, there is a delay in the rate of absorption characterized by a decrease in C max and a delay in time to peak plasma concentration (T max).
Nateglinide did not have any effect on gastric emptying in healthy subjects as assessed by acetaminophen testing.
Distribution
Following intravenous (IV) administration of nateglinide, the steady-state volume of distribution of nateglinide is estimated to be approximately 10 L in healthy subjects. Nateglinide is extensively bound (98%) to serum proteins, primarily serum albumin, and to a lesser extent α 1 acid glycoprotein. The extent of serum protein binding is independent of drug concentration over the test range of 0.1 to 10 mcg/mL.Elimination
In healthy volunteers and patients with type 2 diabetes mellitus, nateglinide plasma concentrations declined with an average elimination half-life of approximately 1.5 hours.Metabolism
In vitro drug metabolism studies indicate that nateglinide is predominantly metabolized by the cytochrome P450 isozyme CYP2C9 (70%) and to a lesser extent CYP3A4 (30%).The major routes of metabolism are hydroxylation followed by glucuronide conjugation. The major metabolites are less potent antidiabetic agents than nateglinide. The isoprene minor metabolite possesses potency similar to that of the parent compound nateglinide.
Excretion
Nateglinide and its metabolites are rapidly and completely eliminated following oral administration. Eighty-three percent of the 14 C-nateglinide was excreted in the urine with an additional 10% eliminated in the feces. Approximately 16% of the 14 C-nateglinide was excreted in the urine as parent compound.Specific Populations
Renal Impairment
No pharmacokinetic data are available in subjects with mild renal impairment (CrCl 60 to 89 mL/min). Compared to healthy matched subjects, patients with type 2 diabetes mellitus and moderate and severe renal impairment (CrCl 15-50 mL/min) not on dialysis displayed similar apparent clearance, AUC, and C max. Patients with type 2 diabetes and renal failure on dialysis exhibited reduced overall drug exposure (C max decreased by 49%; not statistically significant). However, hemodialysis patients also experienced reductions in plasma protein binding compared to the matched healthy volunteers.In a cohort of 8 patients with type 2 diabetes and end-stage renal disease (ESRD) (eGFR < 15 mL/min/1.73m 2) M1 metabolite accumulation up to 1.2 ng/mL occurred with a dosage of 90 mg once daily for 1 to 3 months. In another cohort of 8 patients with type 2 diabetes on hemodialysis, M1 concentration decreased after a single session of hemodialysis. Although the hypoglycemic activity of the M1 metabolite is approximately 5 times lower than nateglinide, metabolite accumulation may increase the hypoglycemic effect of the administered dose.
Hepatic Impairment
In patients with mild hepatic impairment, the mean increase in C max and AUC of nateglinide were 37% and 30% respectively, as compared to healthy matched control subjects. There is no data on pharmacokinetics of nateglinide in patients with moderate-to-severe hepatic impairment.Gender
No clinically significant differences in nateglinide pharmacokinetics were observed between men and women.Race
Results of a population pharmacokinetic analysis including subjects of Caucasian, Black, and other ethnic origins suggest that race has little influence on the pharmacokinetics of nateglinide.Age
Age does not influence the pharmacokinetic properties of nateglinide.Drug Interactions:
In vitro assessment of drug interactions
Nateglinide is a potential inhibitor of the CYP2C9 isoenzyme in vivo as indicated by its ability to inhibit the in vitro metabolism of tolbutamide. Inhibition of CYP3A4 metabolic reactions was not detected in in vitro experiments.In vitro displacement studies with highly protein-bound drugs such as furosemide, propranolol, captopril, nicardipine, pravastatin, glyburide, warfarin, phenytoin, acetylsalicylic acid, tolbutamide, and metformin showed no influence on the extent of nateglinide protein binding. Similarly, nateglinide had no influence on the serum protein binding of propranolol, glyburide, nicardipine, warfarin, phenytoin, acetylsalicylic acid, and tolbutamide in vitro. However, prudent evaluation of individual cases is warranted in the clinical setting.
In vivo assessment of drug interactions
The effect of coadministered drugs on the pharmacokinetics of nateglinide and the effect of nateglinide on pharmacokinetics of coadministered drugs are shown in Tables 3 and 4. No clinically relevant change in pharmacokinetic parameters of either agent was reported when nateglinide was coadministered with glyburide, metformin, digoxin, warfarin, and diclofenac.Table 3: Effect of Coadministered Drugs on Pharmacokinetics of Nateglinide AM: after morning dose; PM: after evening dose; *after second dose; ↑: increase in the parameter; ↓: decrease in the parameter Coadministered drug
Dosing regimen of coadministered drug
Dosing regimen of nateglinide
Change in C max
Change in AUC
Glyburide
10 mg once daily for 3 weeks
120 mg three times a day, single dose
8.78% ↓
3.53 % ↓
Metformin
500 mg three times a day for 3 weeks
120 mg three times a day, single dose
AM: 7.14% ↑
PM: 11.4% ↓
AM: 1.51% ↑
PM: 5.97% ↑
Digoxin
1 mg, single dose
120 mg three times a day, single dose
AM: 2.17% ↓
PM: 3.19% ↑
AM: 7.62% ↑
PM: 2.22% ↑
Warfarin
30 mg, single dose
120 mg three times a day for 4 days
2.65% ↑
3.72% ↓
Diclofenac
75 mg, single dose
120 mg twice daily, single dose
AM: 13.23% ↓
*PM: 3.76% ↑
AM: 2.2% ↓
*PM: 7.5% ↑
Table 4: Effect of Nateglinide on Pharmacokinetics of Coadministered Drugs AM: after morning dose; PM: after evening dose; SD: single dose; ↑: increase in the parameter; ↓: decrease in the parameter Coadministered drug
Dosing regimen of coadministered drug
Dosing regimen of nateglinide
Change in C max
Change in AUC
Glyburide
10 mg once daily for 3 weeks
120 mg three times a day, single dose
3.18% ↓
7.34% ↓
Metformin
500 mg three times a day for 3 weeks
120 mg three times a day, single dose
AM: 10.7% ↑
PM: 0.40% ↑
AM: 13.3% ↑
PM: 2.27% ↓
Digoxin
1 mg, single dose
120 mg three times a day, single dose
5.41% ↓
6.58 % ↑
Warfarin
30 mg, single dose
120 mg three times a day for 4 days
R-warfarin: 1.03% ↓
S-warfarin: 0.85% ↓
R-warfarin: 0.74% ↑
S-warfarin: 7.23% ↑
Diclofenac
75 mg, single dose
120 mg twice daily, single dose
2.19% ↑
7.97% ↑
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity: Nateglinide did not increase tumors in two year carcinogenicity studies conducted in mice and rats. Oral doses of Nateglinide up to 900 mg/kg in rats and 400 mg/kg in mice were tested, which produced exposures in rats approximately 30-40 times and in mice 10-30 times the human therapeutic exposure of nateglinide at a dose of 120 mg three times daily, based on AUC.
Mutagenesis: Nateglinide was not genotoxic in the in vitro Ames test, mouse lymphoma assay, chromosome aberration assay or in the in vivo mouse micronucleus test.
Impairment of Fertility: Fertility was unaffected by administration of nateglinide to rats at doses up to 600 mg/kg (corresponding to 16 times the MRHD of 120 mg three times per day, based on BSA).
-
14 CLINICAL STUDIES
14.1 Monotherapy
In a 24-week, double-blind, placebo-controlled study, patients with type 2 diabetes were randomized to receive either nateglinide (60 mg or 120 mg three times daily before meals) or placebo. Patients previously treated with antidiabetic medications were required to discontinue that medication for at least 2 months before randomization.
At Week 24, treatment with nateglinide before meals resulted in statistically significant reductions in mean HbA 1C and mean fasting plasma glucose (FPG) compared to placebo (see Table 5). The reductions in HbA 1C and FPG were similar for patient's naïve to, and those previously exposed to, antidiabetic medications.
Table 5: Endpoint Results for a 24-week, Fixed Dose Study of Nateglinide Monotherapy - *
- p-value ≤ 0.004
Placebo
Nateglinide 60 mg three times daily before meals
Nateglinide 120 mg three times daily before meals
HbA 1C (%)
N = 168
N = 167
N = 168
Baseline (mean)
8.0
7.9
8.1
Change from baseline (mean)
+0.2
-0.3
-0.5
Difference from placebo (mean)
-0.5 *
-0.7 *
FPG (mg/dL)
N = 172
N = 171
N = 169
Baseline (mean)
167.9
161.0
166.5
Change from baseline (mean)
+9.1
+0.4
-4.5
Difference from placebo (mean)
-8.7 *
-13.6 *
14.2 Monotherapy Compared to Glyburide
In a 24-week, double-blind, active-controlled trial, patients with type 2 diabetes who had been on a sulfonylurea for 3 or more months and who had a baseline HbA 1C greater than or equal to 6.5% were randomized to receive nateglinide (60 mg or 120 mg three times daily before meals) or glyburide 10 mg once daily. Patients randomized to nateglinide had statistically significant increases in mean HbA 1C and mean FPG at endpoint compared to patients randomized to glyburide.
Table 6: Endpoint Results for a 24-Week Study of Nateglinide Monotherapy Compared to Glyburide - *
- p-value < 0.001
Glyburide 10 mg
Once daily
Nateglinide 60 mg
three times daily before meals
Nateglinide 120 mg
three times daily before meals
HbA 1c (%)
N = 183
N = 178
N = 179
Baseline (mean)
7.8
8.0
7.9
Change from baseline (mean)
0.3
1.3
1.1
Difference from glyburide
1.0 *
0.9 *
FPG (mmol/L)
N = 184
N = 182
N = 180
Baseline (mean)
9.44
9.67
9.61
Change from baseline (mean)
0.19
3.06
2.84
Difference from glyburide
2.87 *
2.66 *
14.3 Monotherapy and In Combination with Metformin
In a 24-week, double-blind, active- and placebo-controlled study, patients with type 2 diabetes were randomized to receive either nateglinide alone (120 mg three times daily before meals), metformin alone (500 mg three times daily), a combination of nateglinide 120 mg (three times daily before meals) and metformin (500 mg three times daily), or placebo. Fifty-seven percent of patients were previously untreated with oral antidiabetic therapy. Patients previously treated with antidiabetic medications were required to discontinue medication for at least 2 months before randomization.
At Week 24, statistically significant reductions in mean HbA 1c and FPG were observed with metformin monotherapy compared to nateglinide monotherapy, and the combination of nateglinide and metformin compared to either nateglinide or metformin monotherapy (see Table 7).
Compared to placebo, nateglinide monotherapy was associated with a statistically significant increase in mean body weight, while no significant change in body weight was observed with metformin monotherapy or combination of nateglinide and metformin therapy (see Table 7). Among the subset of patients previously treated with other antidiabetic agents, primarily glyburide, HbA 1C in the nateglinide monotherapy group increased slightly from baseline, whereas HbA 1C was reduced in the metformin monotherapy group (see Table 7).
Table 7: Endpoint Results for a 24-Week Study of Nateglinide Monotherapy and Combination with Metformin Placebo
Nateglinide 120 mg
three times daily before meals
Metformin 500 mg
three times daily
Nateglinide 120 mg
before meals plus Metformin*
HbA 1C (%)
All
N = 160
N = 171
N = 172
N = 162
Baseline (mean)
8.3
8.3
8.4
8.4
Change from baseline (mean)
+0.4
-0.8 ‡
-1.5
Difference from placebo
-0.8 §
-1.2 §
-1.9 §
Naïve
N = 98
N = 99
N = 98
N = 81
Baseline (mean)
8.2
8.1
8.3
8.2
Change from baseline (mean)
+0.3
-0.7 ‡
-0.8 ‡
-1.6
Difference from placebo
-1.0 §
-1.1 §
-1.9 §
Non-Naïve
N = 62
N = 72
N = 74
N = 81
Baseline (mean)
8.3
8.5
8.7
8.7
Change from baseline (mean)
+0.6
-0.8 ‡
-1.4
Difference from placebo
-0.6 §
-1.4 §
-2.0 §
FPG (mg/dL)
All
N = 166
N = 173
N = 174
N = 167
Baseline (mean)
194.0
196.5
196.0
197.7
Change from baseline (mean)
+8.0
-30.0 ‡
-44.9
Difference from placebo
-21.1 §
-38.0 §
-52.9 §
In another 24-week, double-blind, placebo-controlled trial, patients with type 2 diabetes with HbA 1C greater than or equal to 6.8% after treatment with metformin (greater than or equal to 1500 mg daily for at least 1 month) were first entered into a four week run-in period of metformin monotherapy (2000 mg daily) and then randomized to receive either nateglinide (60 mg or 120 mg three times daily before meals) or placebo as add-on to metformin. At the end of treatment, nateglinide 60 mg and 120 mg three times daily resulted in a statistically significantly greater reductions in HbA 1C compared to placebo when added to metformin (-0.4% and -0.6% for nateglinide 60 mg and nateglinide 120 mg plus metformin, respectively).
Table 8: Endpoint Results for a 24-Week Study of Nateglinide Monotherapy as Add-on to Metformin All nateglinide/placebo taken three times daily before meals; all metformin 1000 mg twice daily Placebo + metformin
Nateglinide 60 mg + metformin
Nateglinide 120 mg + metformin
HbA 1c (%)
N = 150
N = 152
N = 154
Baseline (mean)
8.2
8.0
8.2
Change from baseline (mean)
0.01
-0.4
-0.6
Difference from metformin
-0.4 *
-0.6 †
14.4 Add-On Combination Therapy with Rosiglitazone
A 24-week, double blind, multicenter, placebo-controlled trial was performed in patients with type 2 diabetes not adequately controlled on rosiglitazone 8 mg daily. The addition of nateglinide (120 mg three times per day with meals) was associated with statistically significantly greater reductions in HbA 1C compared to placebo as add-on to rosiglitazone. The mean change in weight from baseline was +3 kg for patients treated with nateglinide compared to +1 kg for patients treated with placebo when added to rosiglitazone.
Table 9: Endpoint Results for a 24-Week Study of the Effect of Adding Nateglinide or Placebo to Rosiglitazone - *
- p-value ≤ 0.0001
Placebo +
rosiglitazone 8 mg once daily
Nateglinide 120 mg before meals +
rosiglitazone 8 mg once daily
HbA 1c (%)
N = 191
N = 194
Baseline (mean)
8.4
8.3
Change from baseline (mean)
0.03
-0.7
Difference from rosiglitazone (mean)
-0.7 *
14.5 Add-On Combination Therapy with Glyburide
In a 12-week study of patients with type 2 diabetes inadequately controlled on glyburide 10 mg once daily, the addition of nateglinide (60 mg or 120 mg three times daily before meals) did not produce any additional benefit.
Table 10: Endpoint Results for a 12-week Study of the Effect of Adding Nateglinide or Placebo to Glyburide Placebo or nateglinide given 10 minutes prior to breakfast, lunch, and dinner; glyburide given with the breakfast dose of nateglinide or placebo. Placebo +
glyburide 10 mg once daily
Nateglinide 60 mg before meals +
glyburide 10 mg once daily
Nateglinide 120 mg before meals +
glyburide 10 mg once daily
HbA 1c (%)
N = 58
N = 55
N = 54
Baseline (mean)
8.7
8.7
8.7
Change from baseline (mean)
0.3
0.2
-0.02
Difference from glyburide (mean)
-0.1 *
-0.3 †
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Nateglinide Tablets, USP are supplied in the following package and dose strength forms:
60 mg
Pink color coated, round biconvex, beveled edge tablet debossed with "P 984" on one side and plain on the other side.
Unit dose packages of 30 (3 x 10) NDC 60687-673-21120 mg
Orange color coated, oval shaped biconvex, tablet debossed with "P 985" on one side and plain on the other side.
Unit dose packages of 30 (3 x 10) NDC 60687-684-21Storage and Handling
Store at 25°C (77°F); excursions permitted to 15°C-30°C (59°F-86°F). [See USP Controlled Room Temperature]FOR YOUR PROTECTION: Do not use if blister is torn or broken.
-
17 PATIENT COUNSELING INFORMATION
Administration
Instruct patients to take nateglinide 1 to 30 minutes before meals. Instruct patients that skip meals to skip their dose of nateglinide [see Dosage and Administration (2)].Hypoglycemia
Inform patients that nateglinide can cause hypoglycemia and instruct patients and their caregivers on self-management procedures, including glucose monitoring and management of hypoglycemia. Inform patients that their ability to concentrate and react may be impaired as a result of hypoglycemia. In patients at higher risk for hypoglycemia and patients who have reduced symptomatic awareness of hypoglycemia, increased frequency of blood glucose monitoring is recommended [see Warnings and Precautions (5.1)].Lactation
Advise patients that use of nateglinide is not recommended while breastfeeding [see Use in Specific Populations (8.2)].Drug Interactions
Discuss potential drug interactions with patients and inform them of potential drug-drug interactions with nateglinide. -
PACKAGING INFORMATION
American Health Packaging unit dose blisters (see How Supplied section) contain drug product from Strides Pharma Inc. as follows:
(60 mg / 30 UD) NDC 60687-673-21 packaged from NDC 64380-167
(120 mg / 30 UD) NDC 60687-684-21 packaged from NDC 64380-168Distributed by:
American Health Packaging
Columbus, OH 432178467321/1022F
-
Package/Label Display Panel – Carton – 60 mg

NDC 60687- 673-21
Nateglinide
Tablets, USP60 mg
30 Tablets (3 x 10) Rx Only
Each Tablet Contains:
Nateglinide, USP...................................................................... 60 mgUsual Dosage: See package insert for full prescribing
information.Store at 20° to 25°C (68° to 77°F); excursions permitted between
15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].Keep this and all drugs out of reach of children.
FOR YOUR PROTECTION: Do not use if blister is torn or broken.
The drug product contained in this package is from
NDC # 64380-167, Strides Pharma Inc.Distributed by:
American Health Packaging
Columbus, Ohio 43217767321
0467321/0322 - Package/Label Display Panel – Blister – 60 mg
-
Package/Label Display Panel

NDC 60687- 684-21
Nateglinide
Tablets, USP120 mg
30 Tablets (3 x 10) Rx Only
Each Tablet Contains:
Nateglinide, USP....................................................................120 mgUsual Dosage: See package insert for full prescribing
information.Store at 20° to 25°C (68° to 77°F); excursions permitted between
15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].Keep this and all drugs out of reach of children.
FOR YOUR PROTECTION: Do not use if blister is torn or broken.
The drug product contained in this package is from
NDC # 64380-168, Strides Pharma Inc.Distributed by:
American Health Packaging
Columbus, Ohio 43217768421
0468421/0322 - Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
NATEGLINIDE
nateglinide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:60687-673(NDC:64380-167) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NATEGLINIDE (UNII: 41X3PWK4O2) (NATEGLINIDE - UNII:41X3PWK4O2) NATEGLINIDE 60 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) STARCH, CORN (UNII: O8232NY3SJ) FERRIC OXIDE RED (UNII: 1K09F3G675) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color pink Score no score Shape ROUND (biconvex) Size 1mm Flavor Imprint Code P;984 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:60687-673-21 30 in 1 CARTON 01/12/2023 1 NDC:60687-673-11 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077463 01/12/2023 NATEGLINIDE
nateglinide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:60687-684(NDC:64380-168) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NATEGLINIDE (UNII: 41X3PWK4O2) (NATEGLINIDE - UNII:41X3PWK4O2) NATEGLINIDE 120 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) STARCH, CORN (UNII: O8232NY3SJ) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color orange Score no score Shape OVAL (biconvex) Size 6mm Flavor Imprint Code P;985 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:60687-684-21 30 in 1 CARTON 12/29/2022 1 NDC:60687-684-11 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077463 12/29/2022 Labeler - American Health Packaging (929561009) Establishment Name Address ID/FEI Business Operations American Health Packaging 929561009 repack(60687-673, 60687-684)

