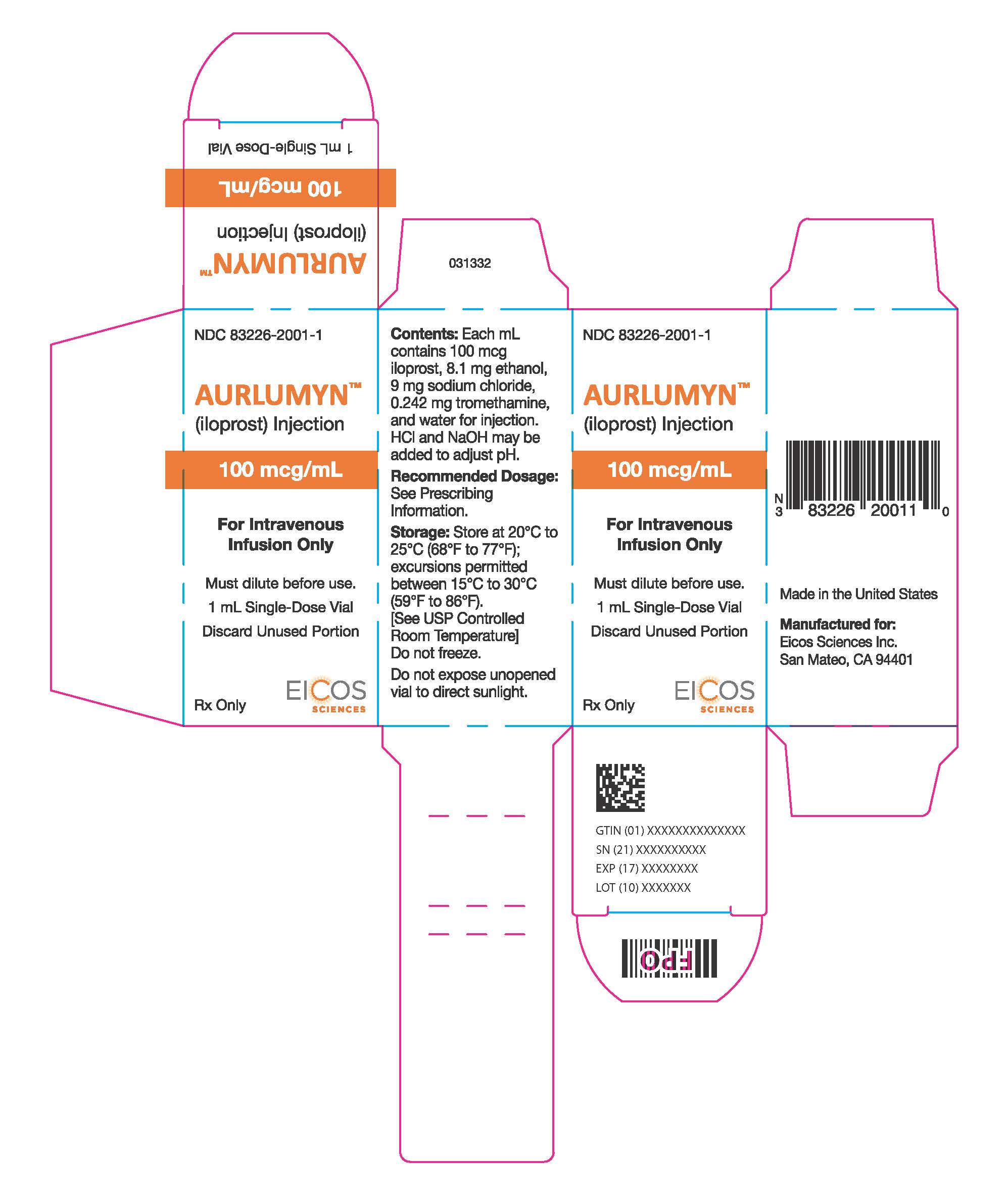
Label: AURLUMYN- iloprost injection, solution

- NDC Code(s): 83226-2001-1
- Packager: Eicos Sciences Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 11, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AURLUMYN safely and effectively. See full prescribing information for AURLUMYN.
AURLUMYN™ (iloprost) injection, for intravenous use
Initial U.S. Approval: 2004INDICATIONS AND USAGE
AURLUMYN is a prostacyclin mimetic indicated for the treatment of severe frostbite in adults to reduce the risk of digit amputations. Effectiveness was established in young, healthy adults who suffered frostbite at high altitudes ( 1.1).
DOSAGE AND ADMINISTRATION
- Initiate intravenous infusion at 0.5 ng/kg/minute and titrate in 0.5 ng/kg/minute increments based on tolerability at intervals of 30 minutes to a maximum of 2 ng/kg/minute ( 2.1).
- Administer as continuous infusion for 6 hours each day up to a maximum of 8 consecutive days ( 2.1).
- Patients with moderate or severe hepatic impairment (Child-Pugh Class B or C): initiate infusion at 0.25 ng/kg/minute and titrate as described above ( 2.3).
- Patients with renal impairment with eGFR less than 30 mL/min: initiate infusion at 0.5 ng/kg/minute and titrate as described above. If the patient cannot tolerate the starting dose of 0.5 ng/kg/minute, the dose can be decreased to 0.25 ng/kg/minute ( 2.4).
- See Full Prescribing Information for instructions on preparation and administration ( 2.2).
DOSAGE FORMS AND STRENGTHS
- Injection: 100 mcg per mL in a single dose vial ( 3).
CONTRAINDICATIONS
- None ( 4).
WARNINGS AND PRECAUTIONS
- AURLUMYN may cause symptomatic hypotension. Monitor vital signs while initiating AURLUMYN. Correct hypotension prior to administration of AURLUMYN ( 5.1).
ADVERSE REACTIONS
Most common adverse events include headache, flushing, palpitations/tachycardia, nausea, vomiting, dizziness, and hypotension ( 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Eicos Sciences, Inc. at 1-844-456-7767 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Frostbite
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Administration
2.3 Use in Patients with Hepatic Impairment
2.4 Use in Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Frostbite
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Monitor vital signs prior to the start of the infusion and with every dose increase.
Administer AURLUMYN as a continuous intravenous infusion over 6 hours each day for up to a maximum of 8 consecutive days.
Start the initial infusion on day 1 at a rate of 0.5 ng/kg/minute and increase in increments of 0.5 ng/kg/minute every 30 minutes according to tolerability up to 2 ng/kg/minute. Dosage is based on actual patient body weight (kg). Repeat dose titration steps on day 2 and day 3. From day 4 onward, start the infusion at the highest tolerated dose from the previous day, and adjust the rate as needed, based on tolerability.
Adverse reactions such as headache, flushing, jaw pain, myalgia, nausea, and vomiting may be dose-limiting. If dose-limiting adverse reactions occur that cannot be tolerated by the patient, then decrease the dose in a stepwise manner by 0.5 ng/kg/min every 30 minutes, until a tolerated dose is reached. If a dose-limiting adverse reaction occurs during administration of AURLUMYN at the starting dose, the infusion should be discontinued, and re-initiation of the infusion can be attempted after the event has resolved or been treated. If infusion is stopped at any point for a dose-limiting adverse event, infusion can be reinitiated at a previously tolerated dose/infusion rate once the event has resolved. The maximum tolerated dose should be maintained for the remaining 6-hour daily infusion.
2.2 Preparation and Administration
Preparation:
Use aseptic technique to prepare AURLUMYN.
Inspect the vial for particulate matter prior to administration. Do not use if the solution is discolored or cloudy or if foreign particles are present.
Dilution:
AURLUMYN should only be diluted using 0.9% Sodium Chloride Injection, USP. Do not dilute or mix AURLUMYN with any other parenteral medications or solutions prior to or during administration.
Withdraw 1 mL (100 mcg) of AURLUMYN solution from the vial and transfer into 100 mL of 0.9% Sodium Chloride Injection, USP polyvinyl chloride (PVC) infusion bag to make a final concentration of 1 mcg/mL (1,000 ng/mL). AURLUMYN can be added to commercially available infusion bags labeled to contain 100 mL of 0.9% Sodium Chloride Injection, USP.
Gently mix the intravenous bag by slowly inverting the bag. Do not shake.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if visibly opaque particles, discoloration, or foreign particles are observed.
Immediately use diluted AURLUMYN infusion solution. If not used immediately, the diluted solution can be stored at room temperature (20°C to 25°C [68°F to 77°F]) for up to 4 hours.
Administration
Administer AURLUMYN as an intravenous infusion through a peripheral line or peripherally inserted central catheter using an infusion pump.
Use an infusion set with an in-line 0.22- or 0.2-micron filter.
Once diluted, AURLUMYN should be administered with an infusion pump that can support the minimum and maximum flow rates. The infusion pump used to administer AURLUMYN should: (1) be able to deliver rates 0.1 to 99.9 mL per hour, (2) adjust infusions rates with increments of 0.1 mL per hour, (3) be accurate to within 5% of programmed rate, and (4) be positive pressure-driven (continuous or pulsatile). The reservoir and infusion line set should be made of polyvinyl chloride.
Infusion rates may be calculated using the following formula:
Infusion Rate (mL/hr) = [Dose (ng/kg/min) × Weight (kg) × 60 min/hr]
Final Concentration (1,000 ng/mL)Avoid inadvertent administration of a bolus of the drug. Do not flush the catheter without withdrawing residual drug from the catheter system.
Discard any unused portion.
2.3 Use in Patients with Hepatic Impairment
Patients with moderate or severe hepatic impairment (Child-Pugh Class B or C): Initiate dosage at 0.25 ng/kg/minute for 30 minutes then continue titration in 0.5 ng/kg/minutes increments every 30 minutes according to tolerability to a maximum dose of 2 ng/kg/minute [see Use in Specific Populations ( 8.6)] .
2.4 Use in Patients with Renal Impairment
Patients with renal impairment with eGFR less than 30 mL/min: Initiate and titrate dosing per recommended dosage. If patient cannot tolerate the starting dose of 0.5 ng/kg/minute the dose can be lowered to 0.25 ng/kg/minute. The effect of dialysis on iloprost exposure has not been evaluated. For patients requiring intermittent hemodialysis, consider iloprost administration after the end of hemodialysis. Alternatively, hemodialysis can be started at least one hour after the end of iloprost infusion.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension
AURLUMYN is a systemic vasodilator and may cause symptomatic hypotension. Correct hypotension prior to administration of AURLUMYN. Monitor vital signs while administering AURLUMYN. Consider temporary discontinuation of concomitant vasodilator or other antihypertensive medications while administering AURLUMYN to reduce potential additive hypotensive effects. Consider down-titration or discontinuation of AURLUMYN if hypotension persists despite discontinuation of other antihypertensives and fluid resuscitation.
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse events reported with the use of intravenous iloprost in patients with frostbite from the published literature include headache, flushing, palpitations/tachycardia, nausea, vomiting, dizziness, and hypotension.
Pre-marketing safety data on AURLUMYN were obtained from 116 patients with Systemic Sclerosis receiving iloprost in 2 multicenter, double-blind, randomized, placebo-controlled studies in patients with Systemic Sclerosis experiencing symptomatic digital ischemic episodes (Raynaud's Phenomenon). Patients received intravenous AURLUMYN administered as a continuous infusion over 6 hours each day for 5 consecutive days and the dose was adjusted according to individual tolerability within the range of 0.5 to 2.0 ng /kg /min. The observed safety profile in these patients was similar to that observed with IV iloprost.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data with AURMULYN during pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are limited published cases of inhaled iloprost use during pregnancy, primarily during the second and third trimesters, that have not identified a drug-associated risk of adverse maternal or fetal outcomes. In animal reproductive studies, administration of continuous intravenous iloprost to pregnant Han-Wistar rats during organogenesis at doses 2-times the maximum recommended human dose on a mg/m 2basis resulted in adverse developmental outcomes. However, there were no adverse developmental outcomes with oral or intravenous administration of iloprost to pregnant Sprague-Dawley rats, rabbits, and monkeys at doses 1111-, 1061-, and 12-times, respectively, the maximum recommended human dose by Cmax ( see Data).
The background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In developmental toxicity studies in pregnant Han-Wistar rats, continuous intravenous administration of iloprost at a dosage of 0.01 mg/kg daily (2 times the maximum recommended human dose on a mg/m 2basis, serum levels not available) led to shortened digits of the thoracic extremity in fetuses and pups. In similar studies in pregnant Sprague-Dawley rats that received iloprost clathrate (13% iloprost by weight) orally at dosages of up to 50 mg/kg/day (C maxof 90 ng/mL), in pregnant rabbits at intravenous dosages of up to 0.5 mg/kg/day (C maxof 86 ng/mL), and in pregnant monkeys at dosages of up to 0.04 mg/kg/day (serum levels of 1 ng/mL), at 1111-, 1062- and 12-times, respectively, the maximum recommended human dose by Cmax, no such digital anomalies or other gross-structural abnormalities were observed in the fetuses/pups. However, in gravid Sprague-Dawley rats, iloprost clathrate (13% iloprost) significantly increased the number of non-viable fetuses at a maternally toxic oral dosage of 250 mg/kg/day (729 times the maximum recommended human dose on a mg/m 2basis) and in Han-Wistar rats was found to be embryolethal in 15 of 44 litters at an intravenous dosage of 1 mg/kg/day (225 times the maximum recommended human dose on a mg/m 2basis).
8.2 Lactation
Risk Summary
There are no data on the presence of iloprost in human milk, the effects on the breastfed infant, or the effects on milk production. Iloprost is present in rat milk ( see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions, advise women not to breastfeed during treatment with iloprost [see Warnings and Precautions ( 5) and Adverse Reactions ( 6)] .
Data
In studies with Han-Wistar rats, higher mortality was observed in pups of lactating dams receiving iloprost intravenously at 1 mg/kg daily (at 225 times the maximum recommended human dose on a mg/m 2basis). In Sprague-Dawley rats, higher mortality was also observed in nursing pups at a maternally toxic oral dose of 250 mg/kg/day of iloprost clathrate (13% iloprost by weight) (729 times the maximum recommended human dose on a mg/m 2basis). In rats, a passage of low levels of iloprost or metabolites into the milk was observed (less than 1% of iloprost dose given intravenously). No disturbance of post-natal development and reproductive performance was seen in animals exposed during lactation.
8.5 Geriatric Use
Clinical studies of iloprost did not include sufficient numbers of subjects aged 65 years and older to determine whether they respond differently than younger subjects. Other reported clinical experience with iloprost has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
In an intravenous iloprost study in patients with liver cirrhosis, the mean clearance in Child-Pugh Class B subjects (n = 5) was approximately 10 mL/min/kg (half that of healthy subjects based on a cross study comparison). In patients with moderate or severe hepatic impairment (Child-Pugh Class B or C), use a lower starting dose of 0.25 ng/kg/minute [see Dosage and Administration ( 2.3)].
8.7 Renal Impairment
In a study of intravenous infusion of iloprost on a dialysis-free day in adults with kidney failure receiving intermittent hemodialysis treatment (n = 7), the mean AUC 0-4hwas 230 pg*h/mL compared to 54 pg*h/mL in patients with kidney failure (n = 8) not requiring intermittent dialysis. The half-life was similar in both groups. The mean AUC 0-4hwas 48 pg*h/mL in normal subjects in a different study. In patients with renal impairment with eGFR less than 30 mL/min, the dose can be lowered to 0.25 ng/kg/minute if the patient cannot tolerate the starting dose of 0.5 ng/kg/minute [see Dosage and Administration ( 2.4)].
The effect of dialysis on the clearance of iloprost has not been evaluated. For patients requiring intermittent hemodialysis, consider iloprost administration after the end of hemodialysis. Alternatively, hemodialysis can be started at least one hour after the end of iloprost infusion.
- 10 OVERDOSAGE
-
11 DESCRIPTION
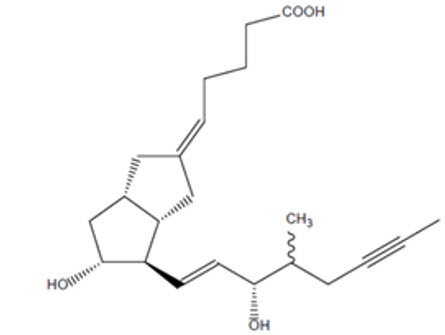
AURLUMYN contains iloprost, a synthetic analog of prostacyclin PGI 2. The chemical name for iloprost is (5 E)-[3a S,4 R,5 R,6a S)-5-hydroxy-4-[(1 E)-(3 S,4 RS)-3-hydroxy-4-methyloct-1-en-6-ynyl]-hexahydropentalen-2(1 H)-ylidene]pentanoic acid. Iloprost consists of a mixture of the 4R and 4S diastereoisomers at a ratio of approximately 53:47. Iloprost is an oily substance, which is soluble in methanol, ethanol, ethyl acetate, acetone, and pH 7 buffer, sparingly soluble in buffer pH 9, and very slightly soluble in distilled water, buffer pH 3, and buffer pH 5. The molecular formula of iloprost is C 22H 32O 4and its molecular weight is 360.49. The structural formula is shown below:
AURLUMYN (iloprost) injection is a clear, colorless, sterile solution formulated for intravenous use. AURLUMYN is supplied in single-use glass vials containing 1-mL per vial. Each mL of the solution contains 100 mcg (0.1 mg) of iloprost as the active ingredient and the following inactive ingredients: 8.1 mg ethanol, 0.9 mg sodium chloride, and 0.242 mg tromethamine. Hydrochloric acid and sodium hydroxide is added to adjust pH to 8.3.
The solution contains no preservatives.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Iloprost is a synthetic analog of prostacyclin PGI 2. Iloprost is a vasodilator and inhibits platelet aggregation.
12.3 Pharmacokinetics
General
Iloprost administered intravenously has linear pharmacokinetics over the dose range of 1 to 3 ng/kg/min. The half-life of iloprost is 20 to 30 minutes.
Absorption and Distribution
Following intravenous infusion, the apparent steady-state volume of distribution was 0.7 to 0.8 L/kg in healthy subjects. Iloprost is approximately 60% protein-bound, mainly to albumin, and this ratio is concentration-independent in the range of 30 to 3000 pg/mL.
Metabolism and Excretion
In vitro studies reveal that cytochrome P450-dependent metabolism plays only a minor role in the biotransformation of iloprost. Iloprost is metabolized principally via β-oxidation of the carboxyl side chain. The main metabolite is tetranor-iloprost, which is found in the urine in free and conjugated form. In animal experiments, tetranor-iloprost was pharmacologically inactive.
Clearance in normal subjects was approximately 20 mL/min/kg.
A mass-balance study using intravenously and orally administered [ 3H]-iloprost in healthy subjects (n = 8) showed recovery of 81% of total radioactivity over 14 hours post-dose, with 68% and 12% recoveries in urine and feces, respectively.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Iloprost was not mutagenic in bacterial and mammalian cells in the presence or absence of extrinsic metabolic activation. Iloprost did not cause chromosomal aberrations in vitro in human lymphocytes and was not clastogenic in vivoin NMRI/SPF mice. There was no evidence of a tumorigenic effect of iloprost clathrate (13% iloprost by weight) in Sprague-Dawley rats dosed orally for up to 8 months at doses of up to 125 mg/kg/day (C maxof 45 ng/mL serum, 556 times the C maxat maximum recommended human dose), followed by 16 months at 100 mg/kg/day , or in Crl:CD-1 ®(ICR)BR albino mice dosed orally for up to 24 months at doses of up to 125 mg/kg/day (C maxof 156 ng/mL serum, 1926 times the Cmax at maximum recommended human dose). The recommended clinical dosage regimen for iloprost (2 ng/kg/min) affords a serum C maxof 0.081 ng/mL. Fertility of males or females was not impaired in Han-Wistar rats at intravenous doses up to 1 mg/kg/day, 225 times the maximum recommended daily dose of Iloprost on a mg/m 2basis.
-
14 CLINICAL STUDIES
14.1 Frostbite
The efficacy of intravenous (IV) iloprost for the treatment of severe frostbite to reduce the risk of digit amputations is derived from a published open-label, randomized controlled trial that enrolled patients with severe frostbite (Cauchy et al, 2011; Cheguillaume, 2011) 1. Severe frostbite was defined as having at least one digit (finger or toe) with frostbite stage 3 (lesion extending just past the proximal phalanx) or stage 4 (lesion extending proximal to the metacarpal or metatarsal joint).
The trial randomized 47 patients at a single site between 1996 and 2008. At enrollment, all eligible patients (n=47) were treated with rapid rewarming of areas with frostbite, aspirin 250 mg IV, and buflomedil 400 mg IV and then randomized to Groups A, B or C. All patients continued to receive aspirin 250 mg IV daily up to 8 days. In addition, Group A (n=15) received buflomedil 400 mg IV for up to 8 days, Group B (n=16) received iloprost IV for 6 hours daily for up to 8 days, and Group C (n=16) received recombinant tissue plasminogen activator IV on Day 1 and iloprost IV for 6 hours daily for up to 8 days.
The mean age of the study population was 33 years (range: 18-55 years), 94% were men, 96% sustained frostbite during sports activities, 6% had history of smoking, and none had diabetes. At randomization, 70% of the patients had frostbite involving the feet, 62% had frostbite involving the hands, and 32% had frostbite involving both feet and hands.
The primary endpoint was the presence of an anomaly (absence of uptake) in the bone phase of technetium 99m scan performed 7 days after initial clinical presentation of frostbite (BS2 bone scintigraphy anomaly) in at least one finger/toe affected by severe frostbite. The BS2 bone scintigraphy anomaly is expected to predict risk of amputation.
On day 7, the presence of BS2 bone scintigraphy anomaly was observed in 60% (9/15), 0% (0/16) and 19% (3/16) of the patients in Groups A, B, and C, respectively. Compared to Group A, the presence of bone scintigraphy anomaly was significantly lower in Group B (p < 0.001) and Group C (p<0.03), favoring iloprost.
Additional follow-up data confirming amputation were obtained for 40/47 patients, which was concordant with the bone scintigraphy results.
- 1
- Cauchy, E., et al. (2011). A controlled trial of a prostacyclin and rt-PA in the treatment of severe frostbite. N Engl J Med 364, 189-190; Cheguillaume, B. (2011). Controlled trial of iloprost and iloprost and rt-PA in the treatment of severe frostbite. Grenoble School of Medicine Thesis. HAL, https://dumas.ccsd.cnrs.fr/dumas-00618697.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
AURLUMYN (iloprost) injection is a clear, colorless sterile solution supplied as 100 mcg per mL single-dose glass vial per carton (NDC 83226-2001-1).
Unopened vials of AURLUMYN are stable until the date indicated on the package when stored at 20°C to 25°C (68°F to 77°F), with excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
The unopened vial should be kept in the carton and not exposed to direct sunlight. Do not freeze.
- 17 PATIENT COUNSELING INFORMATION
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 1 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
AURLUMYN
iloprost injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:83226-2001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ILOPROST (UNII: JED5K35YGL) (ILOPROST - UNII:JED5K35YGL) ILOPROST 100 ug in 1 mL Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) 8.1 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) TROMETHAMINE (UNII: 023C2WHX2V) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:83226-2001-1 1 in 1 CARTON 05/01/2024 1 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217933 05/01/2024 Labeler - Eicos Sciences Inc. (080425839)