Label: XEOMIN- incobotulinumtoxina injection, powder, lyophilized, for solution





-
NDC Code(s):
0259-1605-01,
0259-1610-01,
0259-1620-01,
0259-1620-10, view more0259-4110-01, 0259-4150-01
- Packager: Merz Pharmaceuticals, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated July 17, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XEOMIN® safely and effectively. See full prescribing information for XEOMIN.
XEOMIN (incobotulinumtoxinA) for injection, for intramuscular or intraglandular use
Initial U.S. Approval: 2010WARNING: DISTANT SPREAD OF TOXIN EFFECT
See full prescribing information for complete boxed warning.
The effects of XEOMIN and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults, particularly in those patients who have underlying conditions that would predispose them to these symptoms. (5.1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
XEOMIN is an acetylcholine release inhibitor and neuromuscular blocking agent indicated for the treatment or improvement of:
- Chronic sialorrhea in patients 2 years of age and older (1.1)
- Upper limb spasticity in adults (1.2)
- Upper limb spasticity in pediatric patients 2 to 17 years of age, excluding spasticity caused by cerebral palsy (1.2)
- Cervical dystonia in adults (1.3)
- Blepharospasm in adults (1.4)
- the appearance of upper facial lines in adults:
DOSAGE AND ADMINISTRATION
Chronic Sialorrhea:
- Chronic Sialorrhea in Adults: the recommended total dose is 100 Units per treatment session consisting of 30 Units per parotid gland and 20 Units per submandibular gland, no sooner than every 16 weeks (2.2)
- Chronic Sialorrhea in Pediatric Patients: the recommended dose is based on body weight administered in a 3:2 dose ratio into the parotid and submandibular glands, respectively, no sooner than every 16 weeks; ultrasound guidance recommended (2.2)
Upper limb spasticity, cervical dystonia, and blepharospasm: the optimum dose, frequency, and number of injection sites in the treated muscle(s) should be based on severity and prior treatment response in patients previously treated with botulinum toxin; individualize dosing for each patient:
- Upper Limb Spasticity in Adults: the recommended total dose is up to 400 Units, divided among affected muscles (2.3)
- Upper Limb Spasticity in Pediatric Patients, excluding spasticity caused by cerebral palsy: the recommended total dose is 8 Units/kg (maximum 200 Units) per single upper limb or 16 Units/kg (maximum 400 U) in both upper limbs, divided among affected muscles (2.3)
- Cervical Dystonia: the recommended initial dose is 120 Units per treatment session (2.4)
- Blepharospasm: the recommended initial dose is 50 Units (25 Units per eye) (2.5)
Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines): When treating all three areas simultaneously (glabellar lines, horizontal forehead lines, and lateral canthal lines), the maximum recommended dose is 64 Units (2.6). When not treating simultaneously:
- Glabellar Lines: four Units into each of five sites, for a maximum dose of 20 Units
- Horizontal Forehead Lines treated simultaneously with Glabellar Lines: for HFL four Units into each of five sites (20 Units) and four Units into each of five GL sites (20 Units), for a maximum dose of 40 Units
- Lateral Canthal Lines: four Units into each of three sites per side (six injection sites in total), for a maximum dose of 12 Units per side (24 Units in total)
Administer retreatment with XEOMIN no more frequently than every three months.
Reconstituted XEOMIN:
- Is intended for intramuscular or intraglandular injection in the parotid and submandibular glands only (2.7)
- Use for only one injection session and for only one patient (2.7)
- Instructions are specific for 50 Unit, 100 Unit, and 200 Unit vials (2.7)
- Store in a refrigerator (2°C to 8°C) and use within 24 hours (2.7)
DOSAGE FORMS AND STRENGTHS
- For injection: 50 Units, 100 Units, or 200 Units lyophilized powder in a single-dose vial (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Respiratory, speech, or swallowing difficulties: increased risk if bilateral neck muscle injections are needed, or with pre-existing muscular disorders; immediate medical attention may be required (5.1, 5.4)
- The potency Units of XEOMIN cannot be compared to or converted into Units of any other preparations of botulinum toxin products (5.2)
- Corneal exposure and ulceration: protective measures may be required (5.5)
ADVERSE REACTIONS
The most commonly observed adverse reactions at rates specified below and greater than placebo are:
Chronic Sialorrhea:
- Chronic Sialorrhea in Adults (≥4% of patients): tooth extraction, dry mouth, diarrhea, and hypertension (6.1)
- Chronic Sialorrhea in Pediatric Patients (≥1% of patients): bronchitis, headache, and nausea/vomiting (6.1)
Spasticity:
- Upper Limb Spasticity in Adults (≥2% of patients): seizure, nasopharyngitis, dry mouth, and upper respiratory tract infection (6.1)
- Upper Limb Spasticity in Pediatric Patients (≥3% of patients): nasopharyngitis and bronchitis (6.1)
Cervical Dystonia (≥5% of patients): dysphagia, neck pain, muscle weakness, injection site pain, and musculoskeletal pain (6.1)
Blepharospasm (≥10% of patients): eyelid ptosis, dry eye, visual impairment, and dry mouth (6.1)
Upper facial lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines): (>1% of patients): injection site bruising (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merz Pharmaceuticals, LLC at 888-493-6646 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Aminoglycosides or other agents that interfere with neuromuscular transmission may potentiate the effect of XEOMIN; co-administer only with caution and close observation (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DISTANT SPREAD OF TOXIN EFFECT
1 INDICATIONS AND USAGE
1.1 Chronic Sialorrhea
1.2 Upper Limb Spasticity
1.3 Cervical Dystonia
1.4 Blepharospasm
1.5 Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Safe Use
2.2 Recommended Dose for Chronic Sialorrhea
2.3 Recommended Dose for Upper Limb Spasticity
2.4 Recommended Dose for Cervical Dystonia
2.5 Recommended Dose for Blepharospasm
2.6 Recommended Dose for Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
2.7 Preparation and Reconstitution Technique
2.8 Administration
2.9 Monitoring to Assess Effectiveness
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Spread of Toxin Effect
5.2 Lack of Unit Equivalency Between Botulinum Toxin Products
5.3 Hypersensitivity Reactions
5.4 Dysphagia and Breathing Difficulties
5.5 Corneal Exposure, Corneal Ulceration, and Ectropion in Patients Treated for Blepharospasm
5.6 Risk of Ptosis in Patients Treated for Glabellar Lines
5.7 Human Albumin and Transmission of Viral Diseases
5.8 Pre-existing Conditions at the Injection Site
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Other Agents Interfering with Neuromuscular Transmission
7.2 Anticholinergic Drugs
7.3 Other Botulinum Neurotoxin Products
7.4 Muscle Relaxants
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Chronic Sialorrhea
14.2 Upper Limb Spasticity
14.3 Cervical Dystonia
14.4 Blepharospasm
14.5 Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DISTANT SPREAD OF TOXIN EFFECT
Postmarketing reports indicate that the effects of XEOMIN and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These may include asthenia, generalized muscle weakness, diplopia, blurred vision, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, particularly in those patients who have underlying conditions that would predispose them to these symptoms. In unapproved uses, including lower limb spasticity in children, and in approved indications, cases of spread of effect have been reported at doses comparable to those used to treat cervical dystonia and at lower doses [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
1.1 Chronic Sialorrhea
XEOMIN is indicated for the treatment of chronic sialorrhea in patients 2 years of age and older.
1.2 Upper Limb Spasticity
1.5 Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
XEOMIN is indicated in adult patients for the temporary improvement in the appearance of upper facial lines:
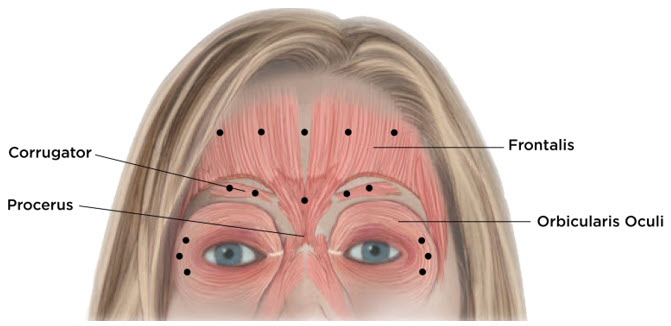
- moderate to severe glabellar lines (GL) associated with corrugator and/or procerus muscle activity
- moderate to severe horizontal forehead lines (HFL) associated with frontalis muscle activity
- moderate to severe lateral canthal lines (LCL) associated with orbicularis oculi muscle activity.
-
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Safe Use
The potency Units of XEOMIN for injection are specific to the preparation and assay method utilized. Units of biological activity of XEOMIN cannot be compared to or converted into Units of any other botulinum toxin products assessed with any other specific assay method [see Warnings and Precautions (5.2) and Description (11)].
Reconstituted XEOMIN is intended for intramuscular or intra-salivary gland injection only.
Do not exceed the recommended maximum cumulative dose in a treatment session for any indication.
2.2 Recommended Dose for Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
The recommended total dose per treatment session is 100 Units. XEOMIN is injected into the parotid and submandibular glands on both sides (i.e., 4 injection sites per treatment session). The recommended total dose per treatment session is 100 Units. The dose is divided with a ratio of 3:2 between the parotid and submandibular glands (Table 1).
Figure 1: Glands for Injection in Chronic Sialorrhea in Adult Patients
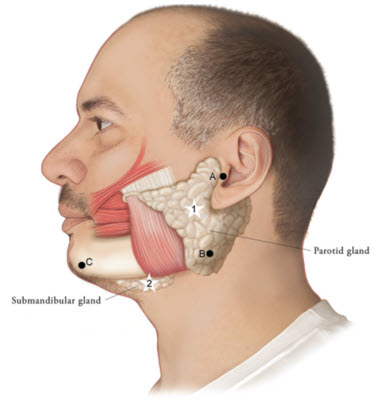
Use the following guidelines if locating salivary glands using anatomic landmarks:
- 1)
- To inject the parotid gland, find the midpoint on the line connecting the tragus and mandible angle (Site A and B, respectively, Figure 1), approximately at the height of the ear lobe. Deliver the injection one finger breadth anterior to this site (Star 1, Figure 1).
- 2)
- To inject the submandibular gland, find the midpoint between the angle of the mandible and the tip of the chin (Site B and C, respectively, Figure 1). Deliver the injection one finger breadth medial to the inferior surface of the mandible at this site (Star 2, Figure 1).
Table 1: Dosing by Gland for Treatment of Chronic Sialorrhea in Adult Patients Gland(s) Units Per Side Total Parotid gland(s) 30 Units 60 Units Submandibular gland(s) 20 Units 40 Units Both Glands 50 Units 100 Units The concentration used in the clinical study after reconstitution was 5 Units/0.1mL. Determine the timing for repeat treatment based on the actual clinical need of the individual patient, and no sooner than every 16 weeks.
Chronic Sialorrhea in Pediatric Patients
XEOMIN is injected into the parotid and submandibular glands on both sides (i.e., 4 injection sites per treatment session). Ultrasound imaging is recommended to guide needle placement into the salivary glands. The body-weight adjusted dose is divided with a ratio of 3:2 between the parotid and submandibular glands (Table 2). XEOMIN has not been studied in children weighing less than 12 kg [see Clinical Studies (14.1)].
Figure 2: Glands for Injection in Chronic Sialorrhea in Pediatric Patients
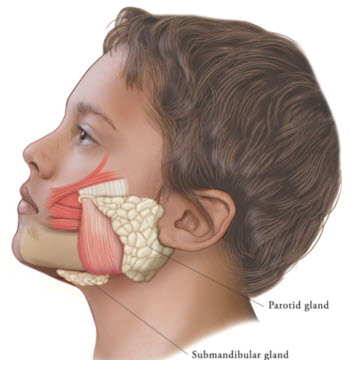
Table 2: Dosing by Body Weight Class for Treatment of Chronic Sialorrhea in Pediatric Patients Body weight Parotid gland, each side Submandibular gland, each side Total dose, both glands, both sides Dose per gland Volume per injection Dose per gland Volume per injection 12 kg or more to less than 15 kg 6 Units 0.24 mL 4 Units 0.16 mL 20 Units 15 kg or more to less than 19 kg 9 Units 0.36 mL 6 Units 0.24 mL 30 Units 19 kg or more to less than 23 kg 12 Units 0.48 mL 8 Units 0.32 mL 40 Units 23 kg or more to less than 27 kg 15 Units 0.6 mL 10 Units 0.4 mL 50 Units 27 kg or more to less than 30 kg 18 Units 0.72 mL 12 Units 0.48 mL 60 Units 30 kg or more 22.5 Units 0.9 mL 15 Units 0.6 mL 75 Units The concentration used in the clinical study after reconstitution was 2.5 Units/0.1 mL. Determine the timing for repeat treatment based on the actual clinical need of the individual patient, and no sooner than every 16 weeks.
2.3 Recommended Dose for Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
Tailor the dosage, frequency, and number of injection sites to the individual patient based on the size, number, and location of muscles to be treated, severity of spasticity, presence of local muscle weakness, patient's response to previous treatment, and adverse event history with XEOMIN. Administer repeat XEOMIN treatments no sooner than every 12 weeks. In patients not previously treated with a botulinum toxin, begin initial dosing at the low end of the recommended dosing range and titrate as clinically necessary. Most patients in clinical studies were retreated between 12 and 14 weeks.
Table 3: XEOMIN Dosing by Muscle for Treatment of Adult Upper Limb Spasticity Clinical Pattern
MuscleUnits (Range) Number of injection sites per muscle Clenched Fist Flexor digitorum superficialis 25 Units-100 Units 2 Flexor digitorum profundus 25 Units-100 Units 2 Flexed Wrist Flexor carpi radialis 25 Units-100 Units 1-2 Flexor carpi ulnaris 20 Units-100 Units 1-2 Flexed Elbow Brachioradialis 25 Units-100 Units 1-3 Biceps 50 Units-200 Units 1-4 Brachialis 25 Units-100 Units 1-2 Pronated Forearm Pronator quadratus 10 Units-50 Units 1 Pronator teres 25 Units-75 Units 1-2 Thumb-in-Palm Flexor pollicis longus 10 Units-50 Units 1 Adductor pollicis 5 Units-30 Units 1 Flexor pollicis brevis/Opponens pollicis 5 Units-30 Units 1 Figure 3: Muscles Involved In Adult Upper Limb Spasticity
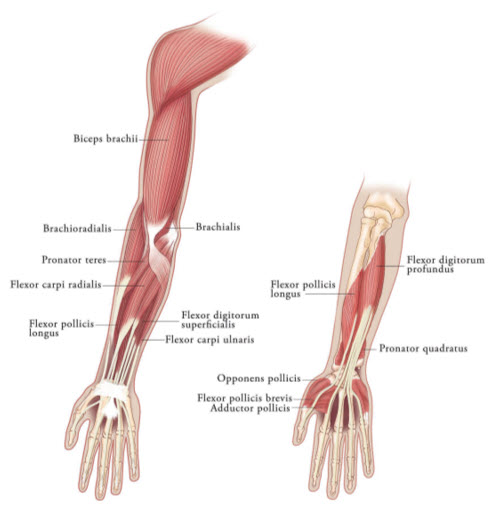
Upper Limb Spasticity in Pediatric Patients, Excluding Spasticity Caused by Cerebral Palsy
Tailor the exact dosage, frequency, and number of injection sites to the individual patient based on size, number and localization of involved muscles; the severity of spasticity; and the presence of local muscle weakness.
The maximum recommended dose is 8 Units/kg, divided among affected muscles, up to a maximum dose of 200 Units per single upper limb. If both upper limbs are treated, do not exceed a total XEOMIN dosage of 16 Units/kg, up to a maximum of 400 Units.
Based on the selected dose, a reconstituted solution at a concentration between 1.25 Units/0.1 mL and 5 Units/0.1 mL is recommended [see Dosage and Administration (2.3)]. Determine the timing for repeat treatment based on the clinical need of the patient; administer repeat treatments no sooner than every 12 weeks. Most patients in clinical studies were retreated between 12 and 16 weeks.
Table 4 includes the recommended dose ranges for the treatment of the clinical patterns of flexed elbow, flexed wrist, pronated forearm, clenched fist, and thumb-in-palm.
Table 4: XEOMIN Dosing by Muscle for Treatment of Pediatric Upper Limb Spasticity, Excluding Spasticity Caused by Cerebral Palsy Clinical Pattern
MuscleDosage Number of Injection Sites per Muscle Range
(Units/kg)Maximum
(Units)Flexed Elbow Brachioradialis 1-2 50 1-2 Biceps 2-3 75 1-3 Brachialis 1-2 50 1-2 Flexed Wrist Flexor carpi radialis 1 25 1 Flexor carpi ulnaris 1 25 1 Pronated Forearm Pronator quadratus 0.5 12.5 1 Pronator teres 1-2 50 1-2 Clenched Fist Flexor digitorum superficialis 1 25 1 Flexor digitorum profundus 1 25 1 Thumb-in-Palm Flexor pollicis longus 1 25 1 Adductor pollicis 0.5 12.5 1 Flexor pollicis brevis/ opponens pollicis 0.5 12.5 1 Figure 4: Muscles Injected for Pediatric Upper Limb Spasticity
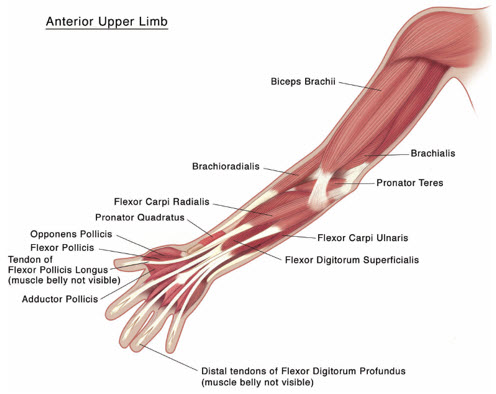
2.4 Recommended Dose for Cervical Dystonia
The recommended initial dose of XEOMIN for cervical dystonia is 120 Units. In a placebo-controlled trial utilizing initial XEOMIN doses of 120 Units and 240 Units, no meaningful difference in effectiveness was demonstrated between the doses [see Clinical Studies (14.3)]. In previously treated patients, their past dose, response to treatment, duration of effect, and adverse event history should be taken into consideration when determining the XEOMIN dose.
In the treatment of cervical dystonia, XEOMIN is usually injected into the sternocleidomastoid, levator scapulae, splenius capitis, scalenus, and/or the trapezius muscle(s) (see Figure 5). This list is not exhaustive, as any of the muscles responsible for controlling head position may require treatment [see Clinical Studies (14.3)]. The dose and number of injection sites in each treated muscle should be individualized based on the number and location of the muscle(s) to be treated, the degree of spasticity/dystonia, muscle mass, body weight, and response to any previous botulinum toxin injections.
The frequency of XEOMIN repeat treatments should be determined by clinical response, but should generally be no more frequent than every 12 weeks [see Clinical Studies (14.3)].
Figure 5: Muscles Involved in Cervical Dsytonia
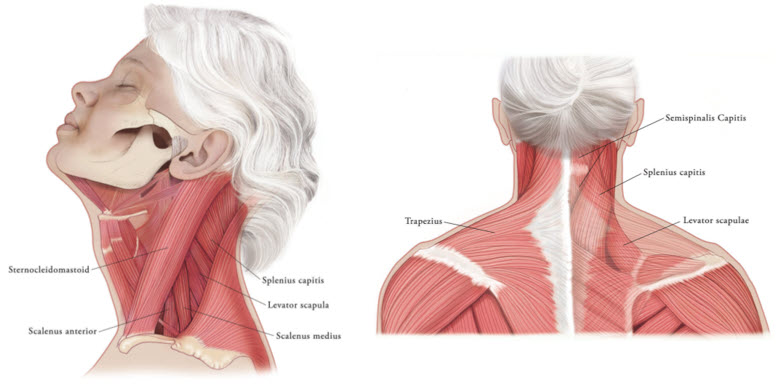
2.5 Recommended Dose for Blepharospasm
In treatment-naïve patients, the recommended initial dose of XEOMIN is 50 Units (25 Units per eye). In patients previously treated with a botulinum toxin A, consider their past dose, response to treatment, duration of effect, and adverse event history when determining the XEOMIN dose.
Do not exceed a total XEOMIN dose of 100 Units per treatment session (50 Units per eye).
Inject XEOMIN into the lateral and medial orbicularis oculi muscle of the upper lid; lateral canthus and the lateral orbicularis oculi muscle of the lower lid; and the corrugator muscle, if necessary (see Figure 6). The number and location of injections may be changed in response to adverse reactions or based on the patient's response to treatment, but do not exceed a total dose of 50 Units per eye.
Figure 6: Injection Sites for Blepharospasm
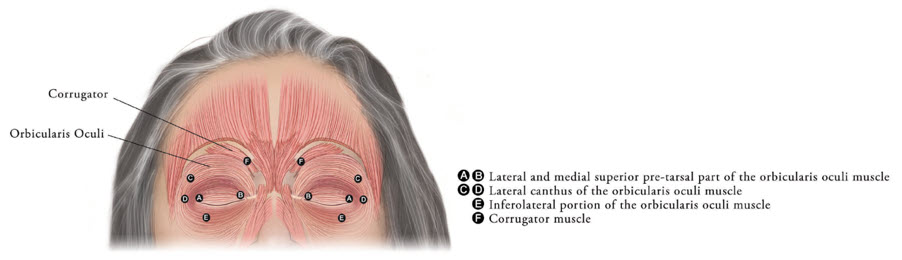
Determine the frequency of XEOMIN repeat treatments by clinical response but administer repeat treatments no more frequent than every 12 weeks [see Clinical Studies (14.4)].
2.6 Recommended Dose for Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
The maximum recommended dose of XEOMIN for simultaneous treatment of upper facial lines [i.e., glabellar lines (GL), horizontal forehead lines (HFL) and lateral canthal lines (LCL)] in adult patients is 64 Units, comprised of 20 Units for GL, 20 Units for HFL, and 24 Units for LCL.
Administer retreatment with XEOMIN no more frequently than every three months.
When not treating upper facial lines (GL, HFL, and LCL) simultaneously in adult patients, refer to the following instructions:
Glabellar Lines
Equally distribute GL treatment to five equal intramuscular injections of 4 Units each. Inject 4 Units of reconstituted XEOMIN intramuscularly into each of 5 sites, 2 in each corrugator muscle and 1 in the procerus muscle for a maximum recommended dose of 20 Units (see Figure 7).
To reduce the complication of ptosis take the following steps:
- Avoid injection near the levator palpebrae superioris, particularly in patients with larger brow depressor complexes.
- Place corrugator injections at least 1 cm above the bony supraorbital ridge.
Horizontal Forehead Lines in Conjunction with Glabellar Lines
Treat HFL in conjunction with GL to minimize the potential for brow ptosis. The maximum recommended dose for treatment of HFL (20 Units) in conjunction with GL (20 Units) is 40 Units.
Equally distribute HFL treatment to 5 horizontally orientated intramuscular injection sites (4 Units each) into the frontalis muscle, at least 2 cm above the orbital rim (see Figure 7).
Lateral Canthal Lines
Inject 4 Units of reconstituted XEOMIN into 3 sites per side (6 total injection sites) in the lateral orbicularis oculi muscle for a total of 12 Units per side (24 Units overall). Place one injection in the horizontal extension of the lateral canthus approximately 1 cm lateral from the bony orbital rim. Place the other two injections approximately 1 cm above and below the area of the first injection (see Figure 7). Give injections with the needle bevel tip up and oriented away from the eye. Avoid injections too close to the zygomaticus major muscle to prevent lip ptosis.
Figure 7: Injection Sites for Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
2.7 Preparation and Reconstitution Technique
Prior to injection, reconstitute each vial of XEOMIN with sterile, preservative-free 0.9% Sodium Chloride Injection, USP [see Dosage Form and Strengths (3)]. A 20-27 gauge short bevel needle is recommended for reconstitution. Draw up an appropriate amount of preservative-free 0.9% Sodium Chloride Injection, USP into a syringe (see Table 5). Clean the exposed portion of the rubber stopper of the vial with alcohol (70%) prior to insertion of the needle. After vertical insertion of the needle through the rubber stopper, the vacuum will draw the saline into the vial. Gently inject any remaining saline into the vial to avoid foam formation. If the vacuum does not pull the saline into the vial, then XEOMIN must be discarded. Remove the syringe from the vial and mix XEOMIN with the saline by carefully swirling and inverting/flipping the vial – do not shake vigorously. Reconstituted XEOMIN is a clear, colorless solution free of particulate matter. Do not use XEOMIN if the reconstituted solution has a cloudy appearance or contains floccular or particulate matter.
After reconstitution, use XEOMIN for only one injection session and for only one patient. Administer reconstituted XEOMIN within 24 hours after dilution. During this time period, store unused reconstituted XEOMIN in the original container in a refrigerator 2°C -8°C (36°F -46°F) for up to 24 hours until time of use. XEOMIN vials are for single-dose only. Discard any unused portion.
Diluent volumes for reconstitution of XEOMIN are indicated in Table 5.
Table 5: Diluent Volumes for Reconstitution of XEOMIN Volume of preservative-free 0.9% Sodium Chloride Injection, USP 50 Unit Vial:
Resulting dose in Units per 0.1 mL100 Unit Vial:
Resulting dose in Units per 0.1 mL200 Unit Vial:
Resulting dose in Units per 0.1 mL- *
- When using 8 mL of diluent for a 100 Unit or 200 Unit vial of XEOMIN, complete the following steps:
- Reconstitute a 100 Unit or 200 Unit vial of XEOMIN with 4 mL of preservative-free 0.9% Sodium Chloride Injection, USP, following instructions above.
- Withdraw 4 mL of preservative-free 0.9% Sodium Chloride Injection, USP, into an appropriately sized syringe for 8 mL in total.
- Using the same syringe, draw up the 4 mL of XEOMIN solution from the reconstituted vial and mix gently.
- †
- When using 16 mL of diluent for a 200 Unit vial of XEOMIN, complete the following steps:
- Reconstitute a 200 Unit vial of XEOMIN with 4 mL of preservative-free 0.9% Sodium Chloride Injection, USP, following instructions above.
- Withdraw 12 mL of preservative-free 0.9% Sodium Chloride Injection, USP, into an appropriately sized syringe for 16 mL in total.
- Using the same syringe, draw up the 4 mL of XEOMIN solution from the reconstituted vial and mix gently.
0.25 mL 20 Units - - 0.5 mL 10 Units 20 Units 40 Units 1 mL 5 Units 10 Units 20 Units 1.25 mL 4 Units 8 Units 16 Units 2 mL 2.5 Units 5 Units 10 Units 2.5 mL 2 Units 4 Units 8 Units 4 mL 1.25 Units 2.5 Units 5 Units 5 mL 1 Unit 2 Units 4 Units 8 mL* - 1.25 Units 2.5 Units 16 mL† - - 1.25 Units 2.8 Administration
Reconstituted XEOMIN is intended for intramuscular or intra-salivary gland injection only.
If proposed injection sites are marked with a pen, DO NOT inject XEOMIN through the pen marks; otherwise a permanent tattooing effect may occur.
For intramuscular injections, the number of injection sites is dependent upon the size of the muscle to be treated and the volume of reconstituted XEOMIN injected.
Inject XEOMIN carefully when injected at sites close to sensitive structures, such as the carotid artery, lung apices, and esophagus. Before administering XEOMIN, the healthcare provider should be familiar with the patient's anatomy and any anatomic alterations, e.g., due to prior surgical procedures.
Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
Use a sterile needle (e.g., 27-30 gauge (0.30-0.40 mm diameter), 12.5 mm length) for intra-salivary gland administration for the treatment of chronic sialorrhea. Inject XEOMIN close to the center of the gland.
The salivary glands can be located using ultrasound imaging or surface anatomical landmarks [see Dosage and Administration (2.3)].
Chronic Sialorrhea in Pediatric Patients
Use a sterile needle (e.g., 27-30 gauge (0.30-0.40 mm diameter), 12.5 mm length) for intra-salivary gland administration for the treatment of chronic sialorrhea. Inject XEOMIN close to the center of the gland.
Ultrasound guidance is recommended for the localization of the involved salivary glands [see Clinical Studies (14.2)].
Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
Use a sterile needle (e.g., 26-gauge (0.45 mm diameter), 37 mm length for superficial muscles; or 22-gauge (0.70 mm diameter), 75 mm length for deeper musculature) in the intramuscular administration in the treatment of upper limb spasticity in adults.
Localization of the involved muscles with electromyographic guidance, nerve stimulation, or ultrasound techniques is recommended.
Upper Limb Spasticity in Pediatric Patients, Excluding Spasticity Caused by Cerebral Palsy
Use a sterile needle (e.g., 30-gauge (0.30 mm diameter), 25 mm length for superficial muscles; or 27-gauge (0.40 mm diameter), 37 mm length for deeper musculature) in the intramuscular administration in the treatment of upper limb spasticity in pediatric patients.
Localization of the involved muscles with techniques such as electromyographic guidance, nerve stimulation, or ultrasound is recommended.
Cervical Dystonia
Use a sterile needle (e.g., 26-gauge (0.45 mm diameter), 37 mm length for superficial muscles; or 22-gauge (0.70 mm diameter), 75 mm length for deeper musculature) in the intramuscular administration in the treatment of cervical dystonia.
Localization of the involved muscles with electromyographic guidance, ultrasound, or nerve stimulation techniques may be useful.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
XEOMIN is contraindicated in patients with:
- Known hypersensitivity to any botulinum toxin product or to any of the components in the formulation [see Warnings and Precautions (5.3) and Description (11)].
- Infection at the proposed injection site(s) because it could lead to severe local or disseminated infection.
-
5 WARNINGS AND PRECAUTIONS
5.1 Spread of Toxin Effect
Postmarketing safety data from XEOMIN and other approved botulinum toxins suggest that botulinum toxin effects may, in some cases, be observed beyond the site of local injection. The symptoms are consistent with the mechanism of action of botulinum toxin and may include asthenia, generalized muscle weakness, diplopia, blurred vision, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence, and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death related to the spread of toxin effects. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can occur in adults treated for spasticity and other conditions, and particularly in those patients who have underlying conditions that would predispose them to these symptoms. In unapproved uses, including lower limb spasticity in children, and in approved indications, symptoms consistent with spread of toxin effect have been reported at doses comparable to or lower than doses used to treat cervical dystonia.
Patients or caregivers should be advised to seek immediate medical care if swallowing, speech, or respiratory disorders occur.
5.2 Lack of Unit Equivalency Between Botulinum Toxin Products
The potency Units of XEOMIN are specific to the preparation and assay method utilized. Units of biological activity of XEOMIN cannot be compared to or converted into Units of any other botulinum toxin products assessed with any other specific assay method [see Description (11)].
5.3 Hypersensitivity Reactions
Serious hypersensitivity reactions have been reported with botulinum toxin products. Hypersensitivity reactions include anaphylaxis, serum sickness, urticaria, soft tissue edema, and dyspnea. If serious and/or immediate hypersensitivity reactions occur, discontinue further injection of XEOMIN and institute appropriate medical therapy immediately. The use of XEOMIN in patients with a known hypersensitivity to any botulinum neurotoxin or to any of the excipients (human albumin, sucrose), could lead to a life-threatening allergic reaction [see Contraindications (4)].
5.4 Dysphagia and Breathing Difficulties
Treatment with XEOMIN and other botulinum toxin products can result in swallowing or breathing difficulties. Patients with pre-existing swallowing or breathing difficulties may be more susceptible to these complications. In most cases, this is a consequence of weakening of muscles in the area of injection that are involved in breathing or swallowing. When distant effects occur, additional respiratory muscles may be involved [see Warnings and Precautions (5.4)].
Deaths as a complication of severe dysphagia have been reported after treatment with botulinum toxin. Dysphagia may persist for several months, and require use of a feeding tube to maintain adequate nutrition and hydration. Aspiration may result from severe dysphagia, and is a particular risk when treating patients in whom swallowing or respiratory function is already compromised.
Treatment of cervical dystonia with botulinum toxins may weaken neck muscles that serve as accessory muscles of ventilation. This may result in critical loss of breathing capacity in patients with respiratory disorders who may have become dependent upon these accessory muscles. There have been post-marketing reports of serious breathing difficulties, including respiratory failure, in patients with cervical dystonia treated with botulinum toxin products.
Patients with smaller neck muscle mass and patients who require bilateral injections into the sternocleidomastoid muscles have been reported to be at greater risk of dysphagia. In general, limiting the dose injected into the sternocleidomastoid muscle may decrease the occurrence of dysphagia.
Patients treated with botulinum toxin may require immediate medical attention should they develop problems with swallowing, speech or respiratory disorders. These reactions can occur within hours to weeks after injection with botulinum toxin [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Patients with neuromuscular disorders with peripheral motor neuropathic diseases, amyotrophic lateral sclerosis, or neuromuscular junctional disorders (e.g., myasthenia gravis or Lambert-Eaton syndrome) may be at increased risk for severe dysphagia and respiratory compromise from typical doses of XEOMIN.
5.5 Corneal Exposure, Corneal Ulceration, and Ectropion in Patients Treated for Blepharospasm
Reduced blinking from injection of botulinum toxin products in the orbicularis muscle can lead to corneal exposure, persistent epithelial defect, and corneal ulceration, especially in patients with VII nerve disorders. As patients with previous eye surgery may have reduced corneal sensation, carefully assess corneal sensation before treatment. Vigorous treatment of any corneal epithelial defect should be employed. This may require protective drops, ointment, therapeutic soft contact lenses, or closure of the eye by patching or other means. Because of its anticholinergic effects, XEOMIN should be used with caution in patients at risk of developing narrow angle glaucoma. To decrease the risk for ectropion, XEOMIN should not be injected into the medial lower eyelid area.
Ecchymosis easily occurs in the soft tissues of the eyelid. Immediate gentle pressure at the injection site can limit the size.
5.6 Risk of Ptosis in Patients Treated for Glabellar Lines
Do not exceed the recommended dosage and frequency of administration of XEOMIN.
In order to reduce the complication of ptosis the following steps should be taken:
- Avoid injection near the levator palpebrae superioris, particularly in patients with larger brow depressor complexes.
- Corrugator injections should be placed at least 1 cm above the bony supraorbital ridge.
5.7 Human Albumin and Transmission of Viral Diseases
This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases and variant Creutzfeldt-Jakob disease (vCJD). There is a theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD), but if that risk actually exists, the risk of transmission would also be considered extremely remote. No cases of transmission of viral diseases, CJD, or vCJD have ever been identified for licensed albumin or albumin contained in other licensed products.
5.8 Pre-existing Conditions at the Injection Site
Use caution when XEOMIN is used where the targeted muscle shows excessive weakness or atrophy.
Use caution when XEOMIN is used in patients who have marked facial asymmetry, with surgical alterations to the facial anatomy, pre-existing eyelid or eyebrow ptosis, when excessive weakness or atrophy is present in the target muscles, excessive dermatochalasis, deep dermal scarring, thick sebaceous skin (e.g., the inability to substantially lessen glabellar lines even by physically spreading them apart).
-
6 ADVERSE REACTIONS
The following adverse reactions to XEOMIN are discussed in greater detail in other sections of the labeling:
- Spread of Effects from Toxin [see Warnings and Precautions (5.1)]
- Lack of Unit Equivalency between Botulinum Toxin Products [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Dysphagia and Breathing Difficulties [see Warnings and Precautions (5.4)]
- Corneal Exposure, Corneal Ulceration, and Ectropion in Patients Treated with XEOMIN for Blepharospasm [see Warnings and Precautions (5.5)]
- Risk of Ptosis in Patients Treated for Glabellar Lines [see Warnings and Precautions (5.6)]
- Human Albumin and Transmission of Viral Diseases [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
Table 6 lists the adverse reactions that occurred in ≥3% of XEOMIN-treated patients in the double-blind, placebo-controlled phase of the study in adult patients with chronic sialorrhea [see Clinical Studies (14.1)]. The most common adverse reactions (≥4%) were tooth extraction, dry mouth, diarrhea, and hypertension. In the controlled portion of this study, 74 patients received 100 Units of XEOMIN, and 36 patients received placebo. XEOMIN-treated patients were 21-80 years old (mean 65 years), and were predominantly male (71%) and White (99.5%).
Table 6: Adverse Reactions (≥3%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Adult Chronic Sialorrhea Study Adverse Reaction XEOMIN 100 Units
(N = 74)
%Placebo
(N = 36)
%Tooth extraction 5 0 Dry mouth 4 0 Diarrhea 4 3 Hypertension 4 3 Fall 3 0 Bronchitis 3 0 Dysphonia 3 0 Back pain 3 0 Dry eye 3 0 Chronic Sialorrhea in Pediatric Patients
Table 7 lists the adverse reactions that occurred in ≥1% of XEOMIN-treated patients 6-17 years of age in the double-blind, placebo-controlled portion of the study in pediatric patients with chronic sialorrhea [see Clinical Studies (14.1)]. Of the patients 6-17 years of age, 148 patients received a dose of XEOMIN according to body weight, and 72 patients received placebo. Thirty-five patients 2-5 years of age received an open-label dose of XEOMIN according to body weight. XEOMIN-treated patients were 2-17 years of age (mean 10 years), predominately male (63%) and White (100%).
Table 7: Adverse Reactions (≥1%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Pediatric Chronic Sialorrhea Study Adverse Reaction XEOMIN
(6-17 years)
(N = 148)
%Placebo
(6-17 years)
(N = 72)
%Bronchitis 1 0 Headache 1 0 Nausea/Vomiting 1 0 The most frequently reported adverse reaction in patients ages 2-5 years after XEOMIN injections was nasopharyngitis (6%).
In the open-label extension period, 222 patients 2-17 years of age received up to three additional treatments with XEOMIN every 16±2 weeks. The safety profile of XEOMIN during the open-label extension period was similar to that observed in the double-blind phase of the placebo-controlled pediatric chronic sialorrhea study.
Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
Table 8 lists the adverse reactions that occurred in ≥2% of XEOMIN-treated patients in two placebo-controlled studies in adult patients with upper limb spasticity. Study 1 and Study 2 were both double-blind, placebo-controlled studies, with an open-label extension [see Clinical Studies (14.2)]. In the controlled portion of these studies, 283 patients received ≥120 Units to 400 Units, of which 217 patients received at least 400 Units of XEOMIN, and 182 patients received placebo. XEOMIN-treated patients were 20-79 years of age (mean 56 years), and were predominantly male (58%), and White (84%).
Table 8: Adverse Reactions (≥2%) and Greater for XEOMIN than Placebo: Double-Blind Phase of Placebo-Controlled Adult Upper Limb Spasticity Study 1 and Study 2 Adverse Reaction XEOMIN 400 Units
(N = 217)
%Placebo
(N = 182)
%Seizure 3 0 Nasopharyngitis 2 0 Dry mouth 2 1 Upper respiratory tract infection 2 1 Upper Limb Spasticity in Pediatric Patients
Table 9 lists the adverse reactions that occurred in ≥2% of XEOMIN-treated patients in Study 1 in pediatric patients 2 years of age and older with upper limb spasticity. In the controlled portion of Study 1, 350 patients were randomized to one of three doses of XEOMIN: 87 received 2 Units/kg per affected upper limb, 87 received 6 Units/kg per affected upper limb, and 176 received 8 Units/kg per affected upper limb [see Clinical Studies (14.2)]. XEOMIN-treated patients were 2 to 17 years of age (mean 7 years), 63% were male, and 90% were White.
No relationship between increased dose and increased occurrence of adverse reactions was observed. The most common adverse reactions (≥3% of XEOMIN-treated patients) at the recommended dose of XEOMIN (8 Units/kg) were nasopharyngitis and bronchitis.
Table 9: Adverse Reactions (≥2%) in Patients Treated with XEOMIN 2 Units/kg or 8 Units/kg: Double-Blind Phase of Study 1 in Pediatric Upper Limb Spasticity Adverse Reactions XEOMIN 2 Units/kg
N=87
%XEOMIN 8 Units/kg
N=176
%- *
- Includes pharyngotonsillitis, pharyngitis and tonsillitis
Infections and infestations Nasopharyngitis 6 3 Bronchitis 2 3 Pharyngotonsillitis* 2 2 Upper respiratory tract infection 2 2 Respiratory tract infection viral 1 2 Injury, poisoning and procedural complications Fall 0 2 Musculoskeletal and connective tissue disorders Pain in extremity 0 2 Cervical Dystonia
The data described below reflect exposure to a single intramuscular dose of XEOMIN in a placebo-controlled, Phase 3 trial in patients with cervical dystonia [see Clinical Studies (14.3)]. In this study, 159 patients received XEOMIN (78 were randomized to receive a total dose of 120 Units, and 81 were randomized to receive a total dose of 240 Units). XEOMIN-treated patients were 18 to 79 years old (mean 53 years), and were predominantly female (66%) and Caucasian (91%). At study baseline, approximately 25% had mild, 50% had moderate, and 25% had severe cervical dystonia. Approximately 61% of XEOMIN-treated patients had previously received another botulinum toxin type A product. Table 10 lists adverse reactions that occurred in ≥5% of XEOMIN-treated patients (in any treatment group) and greater than placebo.
Table 10: Adverse Reactions (≥5%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Cervical Dystonia Study Adverse Reaction XEOMIN 120 Units
(N=77)
%XEOMIN 240 Units
(N=82)
%Placebo
(N=74)
%Musculoskeletal and connective tissue disorders 23 32 11 Neck pain 7 15 4 Muscular weakness 7 11 1 Musculoskeletal pain 7 4 1 Gastrointestinal disorders 18 24 4 Dysphagia 13 18 3 Nervous system disorders 16 17 7 General disorders and administration site conditions 16 11 11 Injection site pain 9 4 7 Infections and infestations 14 13 11 Respiratory, thoracic and mediastinal disorders 13 10 3 Blepharospasm
Study 1 was a randomized, double-blind, placebo-controlled study that only included treatment-naïve patients [see Clinical Studies (14.4)]. In the controlled portion, 22 patients received XEOMIN 25 Units, 19 patients received 50 Units, and 20 patients received placebo. XEOMIN-treated patients were 23 to 78 years of age (mean 55 years). Fifty-nine percent of the patients were women, 77% were Asian, and 23% White. No patients withdrew prematurely because of an adverse event. Table 11 lists the adverse reactions that occurred in ≥6% of XEOMIN-treated patients and greater than placebo.
Table 11: Adverse Reactions (≥6%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Blepharospasm Study 1 Adverse Reaction XEOMIN 50 U
(N=19)
%Placebo
(N=20)
%Eye disorders 21 10 Eyelid ptosis 16 0 Study 2 was a double-blind, placebo-controlled, flexible dose study with an open-label extension (OLEX) period. The study only included patients previously treated with onabotulinumtoxinA (Botox) [see Clinical Studies (14.4)]. In the controlled portion, 74 patients received XEOMIN at a mean dose of approximately 33 Units per eye (minimum 10 Units, maximum 50 Units). XEOMIN-treated patients were 22 to 79 years of age (mean 62 years), predominantly female (65%) and Caucasian (60%). Table 12 lists the adverse reactions that occurred in ≥5% of XEOMIN-treated patients and greater than placebo.
Table 12: Adverse Reactions (≥5%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Blepharospasm Study 2 Adverse Reaction XEOMIN
(N=74)
%Placebo
(N=34)
%- *
- including vision blurred
Eye disorders 38 21 Eyelid ptosis 19 9 Dry eye 16 12 Visual impairment* 12 6 Gastrointestinal disorders 30 15 Dry mouth 16 3 Diarrhea 8 0 Infections and infestations 20 15 Nasopharyngitis 5 3 Respiratory tract infection 5 3 Nervous system disorders 14 9 Headache 7 3 General disorders and administration site conditions 11 9 Respiratory, thoracic and mediastinal disorders 11 3 Dyspnea 5 3 Upper Facial Lines [Glabellar Lines (GL), Horizontal Forehead Lines (HFL), and Lateral Canthal Lines (LCL)]
In two placebo-controlled trials in 730 adult subjects with upper facial lines (GL, HFL, and LCL), 545 subjects received up to 64 Units of XEOMIN and 185 subjects received placebo. XEOMIN-treated subjects were 19 to 76 years old and were predominantly female (82%). Adverse reactions were reported for 62 of the 545 XEOMIN-treated subjects (11%) and for 14 of the 185 placebo-treated subjects (8%).
The most frequent adverse reactions ≥1% and greater for XEOMIN than placebo are presented in Table 13.
Table 13: Adverse Reactions (≥1%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Upper Facial Lines (GL, HFL, and LCL) Trials Upper Facial Lines
(GL, HFL, and LCL)Adverse Reaction XEOMIN N=545
%Placebo N=185
%Injection site bruising 2 1 In the placebo-controlled trials, brow ptosis (0.7%) and injection site discomfort (0.6%) were also reported more frequently for XEOMIN than placebo-treated subjects.
In the two repeated dose upper facial lines (GL, HFL, and LCL) trials with up to three treatments of XEOMIN, adverse reactions were reported for 123 of the 720 subjects (17%). Injection site hematoma was the most common adverse reaction, reported in 8% of subjects, followed by headache (3%), injection site bruising (3%) and brow ptosis (1%). The incidence of these adverse reactions tended to decrease with subsequent treatments.
In both trials, all randomized subjects were followed up for 120 days before they could enter the open-label extension (OLEX) period which comprised two additional treatment cycles, with durations of 120 days each plus up to 30 days for eligibility reassessments per cycle. During OLEX period, eligible subjects received simultaneous upper facial lines injections of XEOMIN at a total dose of 64 U in all three facial areas (20 U in GL, 20 U in HFL, and 24 U in LCL area).
Glabellar Lines
In three placebo-controlled trials in 803 adult subjects with glabellar lines, 535 subjects received a single dose of 20 Units of XEOMIN and 268 subjects received placebo. XEOMIN-treated subjects were 24 to 74 years old, and were predominantly female (88%).
The most frequent adverse reactions ≥1% and greater for XEOMIN than Placebo are presented in Table 14.
Table 14: Adverse Reactions (≥1%) and Greater for XEOMIN than Placebo: Double-Blind Phase of the Placebo-Controlled Glabellar Lines Trials Adverse Reaction XEOMIN
N=535
%Placebo
N=263
%Nervous system disorders Headache 5 2 In the placebo-controlled trials, facial paresis (0.7%), injection site hematoma (0.6%) and eyelid edema (0.4%) were also reported more frequently for XEOMIN than placebo-treated subjects.
In open-label, multiple-dose trials, adverse reactions were reported for 105 of the 800 subjects (13%). Headache was the most common adverse reaction, reported in 7% of subjects, followed by injection site hematoma (1%). Adverse reactions reported in less than 1% of subjects were: facial paresis (brow ptosis), muscle disorder (elevation of eyebrow), injection site pain, and eyelid edema.
6.2 Postmarketing Experience
The following adverse reactions have been reported during post-approval use of XEOMIN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: allergic dermatitis, dysarthria, dysphagia, eye swelling, eyelid edema, flu-like symptoms, herpes zoster, hypersensitivity, injection site pain, injection site reaction, localized allergic reactions (e.g., swelling, edema, erythema, pruritus or rash), muscle spasm, muscular weakness, myalgia, nausea, and persistent dry mouth (> 110 days).
-
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Other Agents Interfering with Neuromuscular Transmission
Co-administration of XEOMIN and aminoglycosides or other agents interfering with neuromuscular transmission (e.g., tubocurarine-type muscle relaxants) should only be performed with caution as these agents may potentiate the effect of the toxin.
7.2 Anticholinergic Drugs
Use of anticholinergic drugs after administration of XEOMIN may potentiate systemic anticholinergic effects.
7.3 Other Botulinum Neurotoxin Products
The effect of administering different botulinum toxin products at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of XEOMIN in pregnant women. XEOMIN should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. XEOMIN was embryotoxic in rats and increased abortions in rabbits when given at doses higher than the maximum recommended human dose (MRHD) for cervical dystonia (120 Units), on a body weight basis.
In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
When XEOMIN was administered intramuscularly to pregnant rats during organogenesis (3 Units/kg, 10 Units/kg, or 30 Units/kg on gestational days [GDs] 6, 12, and 19; or 7 Units/kg on GDs 6 to 19; or 2 Units/kg, 6 Units/kg, or 18 Units/kg on GDs 6, 9, 12, 16, and 19), decreases in fetal body weight and skeletal ossification were observed at doses that were also maternally toxic. The no-effect level for embryotoxicity in rats was 6 Units/kg (3 times the MRHD for cervical dystonia on a body weight basis). Intramuscular administration to pregnant rabbits during organogenesis (1.25 Units/kg, 2.5 Units/kg, or 5.0 Units/kg on GDs 6, 18, and 28) resulted in an increased rate of abortion at the highest dose, which was also maternally toxic. In rabbits, the no-effect level for increased abortion was 2.5 Units/kg (similar to the MRHD for cervical dystonia on a body weight basis).
8.2 Lactation
Risk Summary
There are no data on the presence of XEOMIN in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for XEOMIN and any potential adverse effects on the breastfed infant from XEOMIN or from the underlying maternal conditions.
8.4 Pediatric Use
The safety and effectiveness of XEOMIN have not been established in pediatric patients for the treatment of lower limb spasticity, cervical dystonia, and blepharospasm, or the temporary improvement in the appearance of upper facial lines:
- moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity
- moderate to severe horizontal forehead lines associated with frontalis muscle activity
- moderate to severe lateral canthal lines associated with orbicularis oculi muscle activity [see Warnings and Precautions (5.1)].
Chronic Sialorrhea in Pediatric Patients
The safety and effectiveness of XEOMIN have been established by evidence from an adequate and well-controlled study of XEOMIN in patients 6 to 17 years of age with chronic sialorrhea [See Clinical Studies (14.1)]. Use of XEOMIN in patients 2 to 5 years of age is supported by the findings of efficacy and safety in patients 6 years and older with chronic sialorrhea, and by safety data in patients 2 to 5 years of age. Safety and effectiveness in pediatric patients below the age of 2 years have not been established [see Warnings and Precautions (5.1)].
Upper Limb Spasticity in Pediatric Patients, Excluding Spasticity Caused by Cerebral Palsy
Safety and effectiveness have been established in pediatric patients 2 to 17 years of age [see Warnings and Precautions (5.1), Adverse Reactions (6.1), and Clinical Studies (14.2)]. The safety and effectiveness of XEOMIN have been established by evidence from adequate and well-controlled studies of XEOMIN in patients 2 to 17 years of age with upper limb spasticity. A pediatric assessment for XEOMIN demonstrates that XEOMIN is safe and effective in another pediatric population. However, XEOMIN is not approved for such patient population due to marketing exclusivity for another botulinum toxin. Safety and effectiveness in pediatric patients below the age of 2 years have not been established [see Warnings and Precautions (5.1)].
Juvenile Animal Toxicity Data
In a study in which juvenile rats received intramuscular injections of Xeomin (0, 5, 10, or 30 Units/kg) every other week from postnatal day 21 for 10 weeks, decreased limb use, decreased body weight gain, skeletal muscle atrophy, and decreased bone growth and density were observed at all doses. Male reproductive organ histopathology (atrophy of the germinal epithelium of the testis, associated with hypospermia) was observed at the mid and high doses, and mating behavior was impaired at the high dose. A no-effect dose for adverse effects on development in juvenile animals was not established. The lowest dose tested (5 Units/kg) is less than the human dose of 400 Units on a body weight (kg) basis.
8.5 Geriatric Use
Chronic Sialorrhea
Of the total number of 184 patients in the placebo-controlled study in chronic sialorrhea in adult patients [see Clinical Studies (14.1)], 107 were 65 years of age and over (46 treated with XEOMIN 100 Units, 44 treated with XEOMIN 75 Units, and 17 received placebo). No differences in safety or effectiveness were observed between older and younger patients. Other clinical studies have not identified differences in responses between older and younger patients, but increased sensitivity in older patients cannot be ruled out.
Upper Limb Spasticity
Of the total number of 283 patients in the placebo-controlled studies in upper limb spasticity in adult patients [see Clinical Studies (14.2)], 118 were 65 years of age and over (70 treated with XEOMIN and 48 received placebo), which included 12 patients 75 years of age and over (7 treated with XEOMIN and 5 received placebo). No overall differences in safety or effectiveness were observed between older and younger adult patients. Other clinical studies have not identified differences in responses between older and younger adult patients, but increased sensitivity in older patients cannot be ruled out.
Cervical Dystonia
Of the total number of 233 patients in the placebo-controlled study in cervical dystonia [see Clinical Studies (14.3)], 29 were 65 years of age and over (19 treated with XEOMIN and 10 received placebo). Of these, ten XEOMIN-treated patients and four placebo-treated patients experienced an adverse event. For patients 65 years of age and over treated with XEOMIN, the most common adverse events were dysphagia (21%) and asthenia (11%).
Blepharospasm
Of the total number of 169 patients in the placebo-controlled studies in blepharospasm [see Clinical Studies (14.4)], 61 were 65 years of age and over (45 treated with XEOMIN and 16 received placebo). No overall difference in effectiveness was observed between older and younger patients.
Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
Of the total number of 547 subjects in the placebo-controlled clinical studies for glabellar lines [see Clinical Studies (14.5)], 21 subjects were 65 years of age and over.
Of the total number of 730 subjects in the placebo-controlled clinical studies for upper facial lines GL, HFL, and LCL [see Clinical Studies (14.5)], 46 subjects were 65 years of age and over.
Clinical trials of XEOMIN showed no increase in the incidence of adverse events related to treatment with XEOMIN in patients 65 years of age and over but the trials did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger adult subjects.
-
10 OVERDOSAGE
Excessive doses of XEOMIN may be expected to produce neuromuscular weakness with a variety of symptoms, particularly when treated intramuscularly. Respiratory support may be required where excessive doses cause paralysis of the respiratory muscles. In the event of overdose, the patient should be medically monitored for symptoms of excessive muscle weakness or muscle paralysis [see Warnings and Precautions (5.1, 5.4)]. Symptomatic treatment may be necessary.
Symptoms of overdose are not likely to be present immediately following injection. Should accidental injection or oral ingestion occur, the person should be medically supervised for several weeks for signs and symptoms of excessive muscle weakness or paralysis.
There is no significant information regarding overdose from clinical studies of XEOMIN.
In the event of overdose, antitoxin raised against botulinum toxin is available from the Centers for Disease Control and Prevention (CDC) in Atlanta, GA. However, the antitoxin will not reverse any botulinum toxin-induced effects already apparent by the time of antitoxin administration. In the event of suspected or actual cases of botulinum toxin poisoning, please contact your local or state Health Department to process a request for antitoxin through the CDC. If you do not receive a response within 30 minutes, please contact the CDC directly at 770-488-7100. More information can be obtained at http://www.cdc.gov/ncidod/srp/drugs/formulary.html#1a.
-
11 DESCRIPTION
The active ingredient of XEOMIN is botulinum toxin type A produced from fermentation of Hall strain Clostridium botulinum serotype A. The botulinum toxin complex is purified from the culture supernatant and then the active ingredient is separated from the proteins (hemagglutinins and non-hemagglutinins) through a series of steps yielding the active neurotoxin with molecular weight of 150 kDa, without accessory proteins. XEOMIN is a sterile white to off-white lyophilized powder intended for intramuscular or intra-salivary gland injection after reconstitution with preservative-free 0.9% Sodium Chloride Injection, USP [see Dosage Forms and Strengths (3)]. One vial of XEOMIN contains 50 Units, 100 Units, or 200 Units of incobotulinumtoxinA, human albumin (1 mg), and sucrose (4.7 mg).
The primary release procedure for XEOMIN uses a cell-based potency assay to determine the potency relative to a reference standard. One Unit corresponds to the median intraperitoneal lethal dose (LD50) in mice. As the method for conducting the assay is specific to XEOMIN, Units of biological activity of XEOMIN cannot be converted into Units of any other botulinum toxin assessed with other specific assays.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
XEOMIN blocks cholinergic transmission at the neuromuscular and salivary neuroglandular junction by inhibiting the release of acetylcholine from peripheral cholinergic nerve endings. This inhibition occurs according to the following sequence: neurotoxin binding to cholinergic nerve terminals, internalization of the neurotoxin into the nerve terminal, translocation of the light-chain part of the molecule into the cytosol of the nerve terminal, and enzymatic cleavage of SNAP25, a presynaptic target protein essential for the release of acetylcholine. In both muscles and glands, impulse transmission is re-established by the formation of new nerve endings.
12.3 Pharmacokinetics
Using currently available analytical technology, it is not possible to detect XEOMIN in the peripheral blood following intramuscular or intraglandular injection at the recommended doses.
12.6 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity.
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of XEOMIN.
Of the 2649 patients treated with XEOMIN in clinical trials [see Clinical Studies (14)], 9 (0.3%) patients were positive for neutralizing antibodies after treatment whose antibody status at baseline was unknown and 4 (0.2%) additional patients developed neutralizing antibodies after treatment. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
Of the 180 patients treated with XEOMIN in the main phase and extension period of the adult chronic sialorrhea clinical trial [see Clinical Studies (14.1)], 1 (0.6%) patient was positive for neutralizing antibodies after treatment. The patient had an antibody status unknown at baseline, and had not received a botulinum toxin treatment in the 12 months prior to enrollment in the study. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Chronic Sialorrhea in Pediatric Patients
Of the 252 patients treated with XEOMIN in the main phase and open-label extension period of the pediatric chronic sialorrhea clinical trial [see Clinical Studies (14.1)], antibody measurements were only performed in patients with body weight of 30 kg or more, resulting in 80 patients tested for antibodies at baseline. Three patients tested positive for neutralizing antibodies at baseline and remained positive at the end of the study. No additional patients developed neutralizing antibodies, and none of the patients demonstrated a secondary lack of treatment response.
Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
Of the 456 patients treated with XEOMIN in the main phase and open-label extension period of the adult upper limb spasticity clinical trials (Study 1 and Study 2) [see Clinical Studies (14.2)], 4 patients were positive for neutralizing antibodies at baseline, and 2 (0.4%) additional patients (with unknown antibody status at baseline) were positive after treatment. Both patients had not received a botulinum toxin treatment in the 12 months prior to enrollment in the studies. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Upper Limb Spasticity in Pediatric Patients
Of the 907 patients treated with XEOMIN in clinical trials for treatment of pediatric spasticity [see Clinical Studies (14.2)], 7 patients were positive for neutralizing antibodies at baseline, and 4 (0.4%) additional patients (with unknown antibody status at baseline) were positive after treatment. All of these patients were treated with onabotulinumtoxinA and/or abobotulinumtoxinA prior to enrollment in the study. Patients who had never received a botulinum toxin treatment did not develop neutralizing antibodies after being treated with XEOMIN. Antibody measurements were not performed in patients with <21 kg body weight. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Cervical Dystonia
Of the 227 patients treated with XEOMIN in the main phase and open-label extension period of the cervical dystonia clinical trial [see Clinical Studies (14.3)], 5 patients were positive for neutralizing antibodies at baseline, 1 (0.4%) patient (with unknown antibody status at baseline) was positive after treatment, and 4 (1.8%) additional patients developed neutralizing antibodies after treatment. All of these patients were pre-treated with onabotulinumtoxinA and/or abobotulinumtoxinA prior to enrollment in the study. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Blepharospasm
Of the 163 patients treated with XEOMIN in the main phase and open-label extension period of the blepharospasm clinical trials (Study 1 and Study 2) [see Clinical Studies (14.4)], 1 (0.6%) patient (with unknown antibody status at baseline) was positive for neutralizing antibodies after treatment. The patient had not received a botulinum toxin treatment in the 12 months prior to enrollment in the studies. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
Glabellar Lines
Of the 464 patients treated with XEOMIN in the main phase and open-label extension period of the glabellar lines clinical trials (GL-1 and GL-2) [see Clinical Studies (14.5)], no patients developed neutralizing antibodies after treatment. No patients demonstrated a secondary lack of treatment response due to neutralizing antibodies.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Impairment of Fertility
In a fertility and early embryonic development study in rabbits, males and females were dosed with XEOMIN (1.25 Units/kg, 2.5 Units/kg, or 3.5 Units/kg) intramuscularly every two weeks for 5 and 3 doses, respectively, beginning 2 weeks prior to mating. No effects on mating or fertility were observed. The highest dose tested is approximately twice the maximum recommended human dose for cervical dystonia (120 Units) on a body weight basis.
-
14 CLINICAL STUDIES
14.1 Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
The efficacy and safety of XEOMIN for the treatment of chronic sialorrhea in adult patients were evaluated in a double-blind, placebo-controlled clinical trial (NCT02091739) that enrolled a total of 184 patients with chronic sialorrhea resulting from Parkinson's disease, atypical parkinsonism, stroke, or traumatic brain injury, that was present for at least three months. Patients with a history of aspiration pneumonia, amyotrophic lateral sclerosis, salivary gland or duct malformation, and gastroesophageal reflux disease were excluded. The study consisted of a 16-week main phase, followed by an extension period of dose-blinded treatment with XEOMIN.
In the main phase, a fixed total dose of XEOMIN (100 Units or 75 Units) or placebo was administered into the parotid and submandibular salivary glands in a 3:2 dose ratio. The co-primary efficacy variables were the change in unstimulated Salivary Flow Rate (uSFR, Table 15) and the change in Global Impression of Change Scale (GICS, Table 16) at Week 4 post-injection. A total of 173 treated patients completed the main phase of the study. For both the uSFR and GICS, XEOMIN 100 Units was significantly better than placebo (see Table 15 and Table 16). XEOMIN 75 Units was not significantly better than placebo.
Table 15: Mean Change in uSFR (g/min) from Baseline at Week 4, 8, 12, and 16 of Main Phase XEOMIN 100 Units Placebo N=73 N=36 - *
- p=0.004
Week 4* -0.13 -0.04 Week 8 -0.13 -0.02 Week 12 -0.12 -0.03 Week 16 -0.11 -0.01 Table 16: Mean GICS at Week 4, 8, 12, and 16 of Main Phase XEOMIN 100 Units Placebo N=74 N=36 - *
- p=0.002
Week 4* 1.25 0.67 Week 8 1.30 0.47 Week 12 1.21 0.56 Week 16 0.93 0.41 In the extension period, patients received up to three additional treatments with XEOMIN 100 Units or 75 Units every 16±2 weeks, for a total exposure duration of up to 64 weeks. Patients had periodic dental examinations to monitor for changes in dentition and oral mucosa. A total of 151 patients completed the extension period.
Chronic Sialorrhea in Pediatric Patients
The efficacy and safety of XEOMIN for the treatment of chronic sialorrhea in pediatric patients were evaluated in a prospective, randomized, double-blind, placebo-controlled (ages 6-17 years), parallel-group, multicenter trial (NCT02270736) that enrolled and treated a total of 216 pediatric patients 6-17 years of age with chronic sialorrhea associated with cerebral palsy, other genetic or congenital disorders, or traumatic brain injury. An additional 35 patients 2-5 years of age were treated with open-label XEOMIN in that study. The study consisted of a 16-week main phase, followed by an open-label extension period of treatment with XEOMIN where patients could receive up to 3 additional treatments with XEOMIN every 16 ± 2 weeks, for a total exposure duration of up to 64 weeks (222 patients completed the extension period).
In the main phase, patients 6-17 years of age were administered a total dose of XEOMIN according to body weight (up to 75 Units), or placebo, into the parotid and submandibular glands in a 3:2 dose ratio, using ultrasound guidance. Patients 2-5 years of age all received open-label treatment with XEOMIN, according to body weight, using ultrasound guidance. Patients with a body weight <12 kg were excluded.
The primary efficacy analysis was conducted in the 6-17 years of age patient group. The co-primary endpoints were the change in unstimulated Salivary Flow Rate (uSFR, Table 17) and carer's Global Impression of Change Scale (GICS, Table 18) at Week 4 post-injection.
For both the uSFR and GICS, XEOMIN was statistically significantly better than placebo (see Table 17 and Table 18).
Table 17: Mean change in uSFR (g/min) from Baseline at Week 4, 8, 12, and 16 of Main Phase XEOMIN
(6-17 years)
N = 148Placebo
(6-17 years)
N=72- *
- p=0.0012
Week 4* -0.14 -0.07 Week 8 -0.16 -0.07 Week 12 -0.16 -0.06 Week 16 -0.15 -0.08 Table 18: Mean carer's GICS at Week 4, 8, 12, and 16 of Main Phase XEOMIN
(6-17 years)
N = 148Placebo
(6-17 years)
N=72- *
- p=0.0320
Week 4* 0.91 0.63 Week 8 0.94 0.54 Week 12 0.87 0.47 Week 16 0.77 0.38 Efficacy in pediatric patients 2 to 5 years of age is extrapolated from the finding of efficacy in older pediatric patients.
14.2 Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
The efficacy and safety of XEOMIN for the treatment of upper limb spasticity in adult patients were evaluated in two Phase 3, randomized, multi-center, double-blind studies.
Study 1 (NCT01392300) and Study 2 (NCT00432666) were both prospective, double-blind, placebo-controlled, randomized, multi-center trials with an open-label extension period (OLEX) to investigate the efficacy and safety of XEOMIN in the treatment of post-stroke spasticity of the upper limb. For patients who had previously received botulinum toxin treatment in any body region, Study 1 and Study 2 required that ≥12 months and ≥4 months, respectively, had passed since the most recent botulinum toxin administration.
Study 1 consisted of a 12-week main phase followed by three 12-week OLEX treatment cycles for a total exposure duration of 48 weeks. The study included 317 treatment-naïve patients who were at least three months post-stroke in the main study period (210 XEOMIN and 107 placebo). During the main period, XEOMIN (fixed total dose of 400 Units) and placebo were administered intramuscularly to the defined primary target clinical pattern chosen from among the flexed elbow, flexed wrist, or clenched fist patterns and to other affected muscle groups. 296 treated patients completed the main phase and participated in the first OLEX cycle. Each OLEX cycle consisted of a single treatment session (XEOMIN 400 Units total dose, distributed among all affected muscles) followed by a 12-week observation period.
Study 2 consisted of a 12-to-20-week main phase followed by an OLEX period of 48 – 69 weeks, for up to 89 weeks of exposure to XEOMIN. The study included 148 treatment-naïve and pre-treated patients with a confirmed diagnosis of post-stroke spasticity of the upper limb who were at least six months post-stroke (73 XEOMIN and 75 placebo). During the main period, for each patient, the clinical patterns of flexed wrist and clenched fist were treated with fixed doses (90 Units and 80 Units, respectively). Additionally, if other upper limb spasticity patterns were present, the elbow, forearm and thumb muscles could be treated with fixed doses of XEOMIN per muscle. 145 patients completed the main phase and participated in the OLEX period, during which time the dosing of each involved muscle could be adapted individually. During the main and OLEX periods, the maximum total dose per treatment session and 12-week interval was 400 Units.
The average XEOMIN doses injected into specific muscles and the number of injection sites per muscle in Study 1 and Study 2 are presented in Table 19.
Table 19: Doses Administered to Individual Muscles (Main Period) in Adult Upper Limb Spasticity Study 1 and Study 2 Intent to Treat (ITT) Muscle Group Muscle Study 1
Units InjectedInjection Site Per Muscle Study 2
Units InjectedInjection Site Per Muscle XEOMIN
(N=210)
Mean±SDXEOMIN
Median
(Min; Max)XEOMIN
(N=73)
Mean±SDXEOMIN
Median
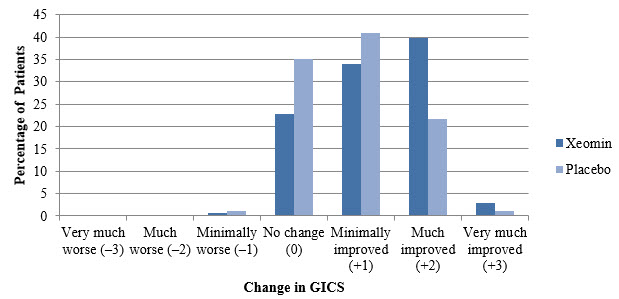
(Min; Max)All Overall 400 ± 2 Units -- 307 ± 77 Units -- Elbow flexors Overall 151 ± 50 Units 5 (1; 11) 142 ± 30 Units 5 (2; 9) Biceps 90 ± 21 Units 3 (1; 4) 80 ± 0 Units 3 (2; 4) Brachialis 52 ± 26 Units 2 (1; 4) 50 ± 0 Units 2 (1; 2) Brachioradialis 43 ± 16 Units 2 (1; 3) 60 ± 2Units 2 (1; 3) Wrist flexors Overall 112 ± 43 Units 4 (1; 6) 90 ± 0 Units 4 (4; 4) Flexor carpi radialis 58 ± 22 Units 2 (1; 3) 50 ± 0 Units 2 (2; 2) Flexor carpi ulnaris 56 ± 22 Units 2 (1; 3) 40 ± 0 Units 2 (2; 2) Finger flexors Overall 104 ± 35 Units 4 (1; 4) 80 ± 0 Units 4 (4; 4) Flexor digitorum profundus 54 ± 19 Units 2 (1; 2) 40 ± 0 Units 2 (2; 2) Flexor digitorum superficialis 54 ± 19 Units 2 (1; 2) 40 ± 0 Units 2 (2; 2) Forearm pronators Overall 52 ± 24 Units 2 (1; 3) 47 ± 16 Units 2 (1; 3) Pronator quadratus 26 ± 13 Units 1 (1; 1) 25 ± 0 Units 1 (1; 1) Pronator teres 42 ± 13 Units 1 (1; 2) 40 ± 0 Units 1.5 (1; 2) Thumb flexors/ adductors Overall 37 ± 25 Units 2 (1; 4) 25 ± 10 Units 1.5 (1; 3) Adductor pollicis 14 ± 8 Units 1 (1; 1) 10 ± 0 Units 1 (1; 1) Flexor pollicis brevis / opponens pollicis 14 ± 9 Units 1 (1; 1) 10 ± 0 Units 1 (1; 1) Flexor pollicis longus 26 ± 16 Units 1 (1; 2) 20 ± 0 Units 1 (1; 1) In Study 1, the primary efficacy variable was the change from baseline in Ashworth Scale (AS) score of the primary target clinical pattern determined by the investigator at the Week 4 visit. The Ashworth Scale is a clinical measure of the severity of spasticity by judging resistance to passive movement. The spasticity of the elbow flexors, wrist flexors, finger flexors, and thumb muscles as well as the forearm pronators was assessed on the 0 to 4-point Ashworth scale at each visit. The co-primary efficacy variable of Study 1 was the Investigator's Global Impression of Change Scales (GICS) after 4 Weeks of treatment with XEOMIN or placebo. The GICS is a global measure of a subject's functional improvement. Investigators were asked to evaluate the subject's global change in spasticity of the upper limb due to treatment, compared to the condition before the last injection. The response was assessed using a 7-point Likert scale that ranges from –3 (very much worse) to +3 (very much improved). XEOMIN was considered to be superior to placebo in Study 1 only if statistical significance was reached in both the AS and GICS variables.
The primary efficacy results are displayed in Table 20.
Table 20: Efficacy Results by Patterns of Spasticity in Adult Upper Limb Spasticity Study 1, Week 4 Mean Change in Ashworth Scale XEOMIN
(N=171)Placebo
(N=88)The analysis is based on Last Observation Carried Forward in the Intent To Treat population. p<0.001 Total Primary Target Clinical Pattern (flexed wrist, flexed elbow, and clenched fist) -0.9 -0.5 A greater percentage of XEOMIN-treated subjects (43%) than placebo-treated subjects (23%) reported 'very much improved' and 'much improved' in their spasticity (see Figure 8).
Figure 8: Investigator's GICS in Adult Upper Limb Spasticity Study 1
Upper Limb Spasticity in Pediatric Patients
Study 1 (NCT02002884) was a prospective, double-blind, dose-response, randomized, multi-center trial with an open-label extension period to evaluate the efficacy and safety of XEOMIN for the treatment of upper limb spasticity in pediatric patients. Study 1 enrolled a total of 350 pediatric patients 2 to 17 years of age with upper limb spasticity in one or both upper limbs. In the double-blind main period of Study 1, patients were randomized to one of three dosages of XEOMIN: 2 Units/kg (maximum 50 Units per upper limb), 6 Units/kg (maximum 150 Units per upper limb); or 8 Units/kg (maximum 200 Units per upper limb). The maximum dose, if both upper limbs were treated, respectively was 4 Units/kg (maximum 100 Units), 12 Units/kg (maximum 300 Units), or 16 Units/kg (maximum 400 Units). For treatment of flexed elbow, injection of biceps brachii was mandatory. The investigator could select 1 of the 2 other muscles contributing to spasticity of elbow flexion (i.e., brachialis and brachioradialis) for injection. For patients needing treatment for a flexed wrist, both the flexor carpi radialis and flexor carpi ulnaris were injected. Study 1 used a dose-response design, in which the two highest dosages of XEOMIN (8 Units/kg and 6 Units/kg) were compared to the lowest dosage (2 Units/kg), which served as control. In the absence of a placebo control, the efficacy of the 2 Units/kg dosage of XEOMIN could not be evaluated in Study 1.
The co-primary efficacy variables in Study 1 were the change from baseline on the Ashworth Scale for the primary clinical target pattern (i.e., elbow flexors or wrist flexors), and the Investigator's Global Impression of Change Scale (GICS), both at Week 4. The GICS is a global measure of a subject's functional improvement based on a 7-point Likert scale that ranges from -3 = very much worse to +3 = very much improved.
As displayed in Table 21, the change from baseline in Ashworth Scale score was significantly greater for patients treated with XEOMIN 8 Units/kg than for patients treated with XEOMIN 2 Units/kg. The difference in GICS score between patients treated with XEOMIN 8 Units/kg and those treated with XEOMIN 2 Units/kg did not reach statistical significance. However, the clinical meaningfulness of the difference in Ashworth Scale score change between patients treated with XEOMIN 8 Units/kg and those treated with XEOMIN 2 Units/kg was established by a responder analysis, in which the proportion of patients with a 1-point change or greater on the Ashworth Scale was examined. In that analysis, 86% of patients treated with XEOMIN 8 Units/kg met the responder definition, compared to 71% of patients treated with XEOMIN 2 Units/kg (nominal p value = 0.0099).
There was no significant difference in change from baseline in Ashworth Scale score, GICS score, or proportion of responders between patients treated with XEOMIN 6 Units/kg and those treated with XEOMIN 2 Units/kg. Therefore, the efficacy of a 6 Units/kg dosage of XEOMIN for the treatment of upper limb spasticity in pediatric patients was not established in Study 1.
Table 21: Ashworth Scale and GICS Efficacy Results in Pediatric Upper Limb Spasticity Study 1, Week 4 XEOMIN
2 Units/kg
(N=87)XEOMIN
8 Units/kg
(N=176)LS = Least Square Mean difference
CI = Confidence Interval- *
- p-value versus low dose group <0.05
Ashworth Scale Mean Change from Baseline at Week 4 -0.9 -1.2 LS Mean Difference versus XEOMIN 2 Units/kg (95% CIs) -- -0.22*
(-0.40, -0.04)GICS Mean at Week 4 1.6 1.7 LS Mean Difference versus XEOMIN 2 Units/kg (95% CIs) -- 0.09
(-0.10, 0.28)14.3 Cervical Dystonia
XEOMIN has been investigated in a randomized, double-blind, placebo-controlled, multicenter trial (NCT00407030) in a total of 233 patients with cervical dystonia. Patients had a clinical diagnosis of predominantly rotational cervical dystonia, with baseline Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) total score ≥20, TWSTRS severity score ≥10, TWSTRS disability score ≥3, and TWSTRS pain score ≥1. For patients who had previously received a botulinum toxin treatment for cervical dystonia, the trial required that ≥10 weeks had passed since the most recent botulinum toxin administration. Patients with swallowing disorders or any significant neuromuscular disease that might interfere with the study were excluded from enrollment. Patients were randomized (1:1:1) to receive a single administration of XEOMIN 240 Units (n=81), XEOMIN 120 Units (n=78), or placebo (n=74). Each patient received a single administration of 4.8 mL of reconstituted study agent (XEOMIN 240 Units, XEOMIN 120 Units, or placebo). The investigator at each site decided which muscles would receive injections of the study agent, the number of injection sites, and the volume at each site. The muscles most frequently injected were the splenius capitis/semispinalis, trapezius, sternocleidomastoid, scalene, and levator scapulae muscles. Table 22 indicates the average XEOMIN dose, and percentage of total dose, injected into specific muscles in the pivotal clinical trial.
Table 22: XEOMIN 120 Units Initial Dose (Units and % of the Total Dose) by Unilateral Muscle Injected During Double Blind Pivotal Phase 3 Study XEOMIN Dose Injected Number of Patients Injected Per Muscle Median XEOMIN Units 75th percentile XEOMIN Units Sternocleidomastoid 63 25 35 Splenius capitis/ Semispinalis capitis 78 48 63 Trapezius 55 25 38 Levator scapulae 49 25 25 Scalenus (medius and anterior) 27 20 25 Most patients received a total of 2-10 injections into the selected muscles. Patients were assessed by telephone at one week post-injection, during clinic visits at Weeks 4 and 8, and then by telephone assessments or clinic visits every two weeks up to Week 20.
The mean age of the study patients was 53 years, 66% of the patients were women, and 91% were White. At study baseline, 61% of patients had previously received a botulinum toxin as treatment for cervical dystonia. The study was completed by 94% of study patients. Three patients discontinued the study prematurely due to adverse events: two patients in the 240 Unit group experienced musculoskeletal pain and muscle weakness, and one patient in the 120 Unit group experienced nausea and dizziness.
The primary efficacy endpoint was the change in the TWSTRS total score from baseline to Week 4 post-injection, in the intent-to-treat (ITT) population, with missing values replaced by the patient's baseline value. TWSTRS evaluates the severity of dystonia, patient-perceived disability from dystonia, and pain, with a range of possible scores from 0 to 85. The mean change in the total TWSTRS score was significantly greater for both XEOMIN groups than for the placebo group (Table 23).
Table 23: Change in TWSTRS Score at Week 4 in Patients with Cervical Dystonia (Double Blind Pivotal Phase 3 Study) TWSTRS Assessment XEOMIN 240 Units
(N=78)XEOMIN 120 Units
(N=81)Placebo
(N =74)SD = Standard Deviation, CI = Confidence Interval. - *
- p-value(s) are from ANCOVA model.
Total TWSTRS at baseline 42.1 42.6 41.8 Total TWSTRS at Week 4 31.2 32.7 39.5 Mean (SD) Change in TWSTRS score from baseline to Week 4 -10.9 (11.7) -9.9 (10.4) -2.2 (7.3) Mean difference from placebo (95% CI) -9.0 (-12.0, -5.9) -7.5 (-10.4, -4.6) p-value* versus placebo <0.001 <0.001 The efficacy of XEOMIN was similar in patients who were botulinum toxin naïve and those who had received botulinum toxin prior to this study.
Examination of age and gender subgroups did not identify differences in response to XEOMIN among these subgroups.
14.4 Blepharospasm
Treatment-Naïve Patients
The efficacy and safety of XEOMIN for the treatment of blepharospasm in treatment-naïve patients were evaluated in Study 1 (NCT01896895), a randomized, double-blind, placebo-controlled, multi-center trial in a total of 61 patients. Patients had a clinical diagnosis of blepharospasm, with a baseline Jankovic Rating Scale (JRS) severity subscore ≥2. Patients were defined as treatment-naïve if at least 12 months had passed since their last botulinum toxin treatment for blepharospasm. During the placebo-controlled phase, a fixed total dose of 25 Units XEOMIN (n=22), 50 Units XEOMIN (n=19), or placebo (n=20) was administered intramuscularly at 6 injection sites per eye (Figure 9). Of the 61 patients randomized, 55 patients completed the placebo-controlled phase. Patients only continued to the open-label extension (OLEX) period if they had a confirmed need for a re-injection by week 20 of the placebo-controlled phase. A total of 39 patients entered and completed the OLEX phase.
The primary efficacy variable was the change from baseline in JRS Severity subscore determined at Week 6 after the injection. The 50 Unit treatment group demonstrated statistically significant improvements compared to placebo, with a difference of -1.2 (p=0.0004). The change from baseline in the JRS Severity subscore for the 25 Unit treatment group 6 weeks after the injection was not statistically significant, with a difference of -0.5 (p=0.1452) compared to placebo (see Figure 9).
Figure 9: Frequency Distribution of Changes from Baseline JRS Severity Subscore at Week 6 for Treatment-Naïve Patients
Pre-Treated Patients
The efficacy and safety of XEOMIN for the treatment of blepharospasm patients pre-treated with onabotulinumtoxinA (Botox) were evaluated in Study 2 (NCT00406367), a randomized, double-blind, placebo-controlled, multi-center trial in a total of 109 patients. Patients had a clinical diagnosis of benign essential blepharospasm, with baseline JRS Severity subscore ≥2, and a stable satisfactory therapeutic response to previous administrations of onabotulinumtoxinA (Botox). At least 10 weeks had to have elapsed since the most recent onabotulinumtoxinA administration. Patients with any significant neuromuscular disease that might interfere with the study were excluded from enrollment. Patients were randomized (2:1) to receive a single administration of XEOMIN (n=75) or placebo (n=34). Each patient in the XEOMIN group received a XEOMIN treatment (dose, volume, dilution, and injection sites per muscle) that was similar to the most recent onabotulinumtoxinA injection sessions prior to study entry. The highest dose permitted in this study was 100 Units (50 Units per eye); the mean XEOMIN dose was 33 Units per eye.
In Table 24 the most frequently injected sites, the median dose per injection site, and the median number (and range) of injection sites per eye are presented.
Table 24: Median Dose and Median Number of Injection Sites per Eye (Blepharospasm) Injection Area Median Units
XEOMINMedian Number of Injection Sites
(Min-Max)Temporal Area 13 2 (1 – 6) Eyebrow Area 5 1 (1 – 4) Upper Lid Area 10 2 (1 – 4) Lower Lid Area 8 2 (1 – 3) Orbital Rim 5 1 (1 – 3) Patients were assessed during clinic visits at Weeks 3 and 6, and then by telephone or at clinic visits every two weeks up to Week 20.
The mean age of the study patients was 62 years, 65% of the patients were women, and 83% were White. The study was completed by 94% of study patients. Approximately one third of patients had other dystonic phenomena; in all but 1% this was limited to facial, cervical, perioral and mandibular muscles. No patients discontinued the study prematurely due to adverse events.
The primary efficacy endpoint was the change in the JRS Severity subscore from baseline to Week 6 post-injection, in the intent-to-treat (ITT) population, with missing values replaced by the patient's most recent value (i.e., last observation carried forward). In the ITT population, the difference between the XEOMIN group and the placebo group in the change of the JRS Severity subscore from baseline to Week 6 was -1.0 (95% CI -1.4; -0.5) points. Comparison of the XEOMIN group to the placebo group was statistically significant at p<0.001.
Figure 10: Frequency Distribution of Changes from Baseline JRS Severity Subscore at Week 6
Examination of age and gender subgroups did not identify substantial differences in response to XEOMIN among these subgroups.
14.5 Upper Facial Lines (Glabellar Lines, Horizontal Forehead Lines, and Lateral Canthal Lines)
Two randomized, double-blind, multi-center, placebo-controlled clinical trials, Trial 1071 (NCT04594213) and Trial 1070 (NCT04622254), were conducted to evaluate XEOMIN for use in the simultaneous intramuscular treatment of upper facial lines (GL, HFL, and LCL). Each trial included an extension period with two additional open-label treatment cycles. In these two trials a total of 730 adult subjects with GL, HFL, and LCL of at least moderate severity at maximum frown as assessed by the investigator and subject were randomized and treated.
In Trial 1071, a total of 362 subjects were randomized and treated. Of the randomized subjects, 179 subjects were treated with XEOMIN in all three treatment areas (receiving 64 U in total: 20 in GL, 20 U in HFL, 24 U in LCL area), 92 subjects were treated with XEOMIN in the GL and HFL areas (receiving 20 U in the GL area and 20 U in HFL area), and 91 subjects were treated with placebo (receiving equal volume of placebo).
In Trial 1070, a total of 368 subjects were randomized and treated. Of the randomized subjects, 184 subjects were treated with XEOMIN in all three treatment area (receiving 64 U in total: 20 in GL, 20 U in HFL, 24 U in LCL area), 90 subjects were treated with XEOMIN in the LCL area (receiving 24 U in LCL area, 12 U per side), and 94 subjects were treated with placebo (receiving equal volume of placebo).
The mean age of the 730 treated and randomized subjects was 46.5 years. The majority of subjects were female (84%) and White (92%). Fifteen percent (15%) of subjects identified as Hispanic or Latino.
For both trials, the severity of upper facial lines (GL, HFL, and LCL) was assessed at maximum contraction using the 5-point photonumerical Merz Aesthetic Scales (MAS; 0=none, 1= mild, 2=moderate, 3=severe, 4=very severe). The MAS assessment was performed independently by both investigators and subjects. The primary timepoint was Day 30 following the first treatment.
For each upper facial area (GL, HFL, or LCL), treatment success was defined as a score of 0 (none) or 1 (mild) and at least two-grade improvement from baseline to Day 30 as rated on the corresponding scale for GL, HFL, and LCL at maximum contraction as assessed by both the investigator and the subject. The percentage of subjects with treatment success in each treatment area is presented in Table 25.
Table 25. Treatment Success at Day 30 in Adults with Upper Facial Lines (GL/HFL/LCL) in Trial 1071 and Trial 1070 Trial 1071 Trial 1070 XEOMIN
(20 Units GL/ 20 Units HFL/ 24 Units LCL)XEOMIN
(20 Units GL/ 20 Units HFL)Placebo XEOMIN
(20 Units GL/ 20 Units HFL/ 24 Units LCL)XEOMIN
(24 Units LCL)Placebo N=179 N=92 N=91 N=184 N=90 N=94 GL 53% 53% 0% 49% - 0% HFL 67% 62% 0% 58% - 0% LCL 53% - 0% 33% 24% 0% Glabellar Lines
Two identically designed randomized, double-blind, multi-center, placebo controlled clinical trials (Studies GL-1 and GL-2) were conducted to evaluate XEOMIN for use in the temporary improvement of moderate to severe glabellar lines. The trials enrolled 547 healthy adult patients with glabellar lines of at least moderate severity at maximum frown. Three hundred sixty six (366) subjects were treated with 20 Units of XEOMIN and 181 subjects received placebo. Subjects were excluded if they had marked ptosis, deep dermal scarring, or an inability to lessen glabellar lines, even by physically spreading them apart. The mean age of trial subjects was 46 years. The majority of subjects were female (86% and 93% in Studies GL-1 and GL-2, respectively), and predominantly White (89% and 65% respectively). The trial subjects received either 20 Units of XEOMIN or an equal amount of placebo. The total dose was delivered in 5 equally divided intramuscular injections of 4 Units each to specific sites (see Figure 7). Subjects were followed up for 120 days.
Investigators and subjects assessed efficacy at maximum frown on Day 30 of treatment using a 4-point scale (0=none, 1=mild, 2=moderate, 3=severe). Composite treatment success was defined as a 2-grade improvement on this scale compared to baseline for both the investigator's and subject's assessments on Day 30. The percentage of subjects with treatment success was greater on the XEOMIN arm than the placebo arm at Day 30 in both trials (see Table 26).
Table 26: Treatment Success at Day 30 (at Least 2 Grades Improvement from Baseline at Maximum Frown) in Adults with GL in Studies GL-1 and GL-2 GL-1 GL-2 XEOMIN
(N=184)Placebo
(N=92)XEOMIN
(N=182)Placebo
(N=89)- *
- Success on both the Investigator and Subject Assessments
Composite Treatment Success* 111 (60%) 0 (0%) 87 (48%) 0 (0%) Investigator Assessment 141 (77%) 0 (0%) 129 (71%) 0 (0%) Subject Assessment 120 (65%) 0 (0%) 101 (55%) 1 (1%) -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
XEOMIN for injection is a sterile white to off-white lyophilized powder supplied in Type 1 borosilicate glass single-dose vials with tamper-proof aluminum seals and bromobutyl rubber closures that are not made with natural rubber latex in the following pack sizes:
Upper Limb Spasticity and Cervical Dystonia
Package XEOMIN 50 Units XEOMIN 100 Units XEOMIN 200 Units Carton with one single-dose vial NDC 0259-1605-01 NDC 0259-1610-01 NDC 0259-1620-01 Storage and Handling
Store unopened vials of XEOMIN at or below 25°C (77°F). Refrigeration of unopened vials is not required. Do not use after the expiration date on the vial. Reconstituted XEOMIN may be stored in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours until time of use [see Dosage and Administration (2.7)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Swallowing, Speaking, or Breathing Difficulties or Other Unusual Symptoms
Advise patients to inform their healthcare provider if they develop any unusual symptoms, including difficulty with swallowing, speaking, or breathing, or if any existing symptom worsens [see Boxed Warning and Warnings and Precautions (5.1, 5.4)]. Inform patients of the risk of aspiration.
Ability to Operate Machinery or Vehicles
Counsel patients that if loss of strength, muscle weakness, blurred vision, or drooping eyelids occur, they should avoid driving a car or engaging in other potentially hazardous activities.
Corneal Exposure, Corneal Ulceration, and Ectropion in Patients Treated for Blepharospasm
Inform patients that injections of XEOMIN may cause reduced blinking or effectiveness of blinking, and that they should seek immediate medical attention if eye pain or irritation occurs following treatment [see Warnings and Precautions (5.5)].
-
SPL UNCLASSIFIED SECTION
Manufactured by:
Merz Pharmaceuticals GmbH
Eckenheimer Landstrasse 100
Frankfurt Germany
U.S. License Number 1830Distributed by:
Merz Pharmaceuticals, LLC
6601 Six Forks Road, Suite 430
Raleigh, NC 27615and
Merz North America, Inc.
4133 Courtney Street, Suite 10
Franksville, WI 53126©2024 Merz Pharmaceuticals, LLC
XEOMIN® is a registered trademark of Merz Pharma GmbH & Co KGaA.
All trademarks are the property of their respective owners.
Patent www.merztherapeutics.com/us/patents/IN00180-02
-
MEDICATION GUIDE
This Medication Guide has been approved by the U. S. Food and Drug Administration. Revised: 7/2024 MEDICATION GUIDE
XEOMIN® (Zeo-min)
(incobotulinumtoxinA)
for injection, for intramuscular or intraglandular useWhat is the most important information I should know about XEOMIN?
XEOMIN may cause serious side effects that can be life-threatening. Call your doctor or get medical help right away if you have any of these problems after treatment with XEOMIN:-
Problems with swallowing, speaking, or breathing. These problems can happen hours to weeks after an injection of XEOMIN if the muscles that you use to breathe and swallow become weak after the injection. Death can happen as a complication if you have severe problems with swallowing or breathing after treatment with XEOMIN.
- People with certain breathing problems may need to use muscles in their neck to help them breathe. These people may be at greater risk for serious breathing problems with XEOMIN.
- Swallowing problems may last for several months. People who cannot swallow well may need a feeding tube to receive food and water. If swallowing problems are severe, food or liquids may go into your lungs. People who already have swallowing or breathing problems before receiving XEOMIN have the highest risk of getting these problems.
- Spread of toxin effects. In some cases, the effect of botulinum toxin may affect areas of the body away from the injection site and cause symptoms of a serious condition called botulism. The symptoms of botulism include:
- loss of strength and muscle weakness all over the body
- double vision
- blurred vision and drooping eyelids
- hoarseness or change or loss of voice
- trouble saying words clearly
- loss of bladder control
- trouble breathing
- trouble swallowing
These symptoms can happen hours to weeks after you receive an injection of XEOMIN
These problems could make it unsafe for you to drive a car or do other dangerous activities. See "What should I avoid while receiving XEOMIN?"What is XEOMIN?
XEOMIN is a prescription medicine:- that is injected into glands that make saliva and is used to treat long-lasting (chronic) drooling (sialorrhea) in adults and in children 2 to 17 years of age.
- that is injected into muscles and used to:
- treat increased muscle stiffness in the arm because of upper limb spasticity in adults.
- treat increased muscle stiffness in the arm in children 2 to 17 years of age with upper limb spasticity, excluding spasticity caused by cerebral palsy.
- treat the abnormal head position and neck pain with cervical dystonia (CD) in adults.
- treat abnormal spasm of the eyelids (blepharospasm) in adults.
- improve the appearance of upper facial lines (glabellar lines, horizontal forehead lines, and lateral canthal lines) (treated simultaneously or individually) for a short period of time (temporary) in adults.
- 2 years of age for the treatment of chronic sialorrhea
- 2 years of age for the treatment of upper limb spasticity
- 18 years of age for the treatment of upper facial lines (glabellar lines, horizontal forehead lines, and lateral canthal lines), cervical dystonia, or blepharospasm
Do not take XEOMIN if you: - are allergic to XEOMIN or any of the ingredients in XEOMIN. See the end of this Medication Guide for a list of ingredients in XEOMIN.
- had an allergic reaction to any other botulinum toxin products
- have a skin infection at the planned injection site.
Before receiving XEOMIN, tell your doctor about all of your medical conditions, including if you: - have a disease that affects your muscles and nerves (such as amyotrophic lateral sclerosis [ALS or Lou Gehrig's disease], myasthenia gravis or Lambert-Eaton syndrome). See "What is the most important information I should know about XEOMIN?"
- have had any side effects from any other botulinum toxin in the past.
- have a breathing problem, such as asthma or emphysema.
- have a history of swallowing problems or inhaling food or fluid into your lungs (aspiration).
- have drooping eyelids.
- have had eye surgery.
- have had surgery on your face.
- are pregnant or plan to become pregnant. It is not known if XEOMIN can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if XEOMIN passes into your breast milk.
Using XEOMIN with certain other medicines may cause serious side effects. Do not start any new medicines until you have told your doctor that you have received XEOMIN in the past. Especially tell your doctor if you:- have received any other botulinum toxin product in the last four months.
- have received injections of botulinum toxin in the past. Be sure your doctor knows exactly which product you received. The dose of XEOMIN may be different from other botulinum toxin products that you have received.
- have recently received an antibiotic by injection or inhalation.
- take muscle relaxants.
- take an allergy or cold medicine.
- take a sleep medicine.
Know the medicines you take. Keep a list of your medicines with you to show your doctor and pharmacist each time you get a new medicine.How will I receive XEOMIN? - XEOMIN is a shot (injection) that your doctor will give you.
- XEOMIN is injected into your affected muscles or glands.
- Your doctor may change your dose of XEOMIN during treatment.
What should I avoid while taking XEOMIN?
XEOMIN may cause loss of strength or general muscle weakness, blurred vision, or drooping eyelids within hours to weeks of taking XEOMIN. If this happens, do not drive a car, operate machinery, or do other dangerous activities. See "What is the most important information I should know about XEOMIN? "What are the possible side effects of XEOMIN?
XEOMIN may cause serious side effects, including:
See "What is the most important information I should know about XEOMIN?"- Injury to the cornea (the clear front surface of the eye) in people treated for blepharospasm. People who receive XEOMIN to treat spasm of the eyelid may have reduced blinking that can cause a sore on their cornea or other problems of the cornea. Call your healthcare provider or get medical care right away if you have eye pain or irritation after treatment with XEOMIN.
- XEOMIN may cause other serious side effects including allergic reactions. Symptoms of an allergic reaction to XEOMIN may include: itching, rash, redness, swelling, wheezing, trouble breathing, or dizziness or feeling faint. Tell your doctor or get medical help right away if you get wheezing or trouble breathing, or if you get dizzy or faint.
- needing to have a tooth pulled (extracted)
- dry mouth
- diarrhea
- high blood pressure
The most common side effects of XEOMIN in children 2 to 17 years of age with chronic sialorrhea include: - bronchitis
- nausea
- headache
- vomiting
The most common side effects of XEOMIN in adults with upper limb spasticity include: - seizure
- nasal congestion, sore throat and runny nose
- dry mouth
- upper respiratory infection
The most common side effects of XEOMIN in children 2 to 17 years of age with upper limb spasticity include: - nasal congestion, sore throat, and runny nose
- bronchitis
The most common side effects of XEOMIN in adults with cervical dystonia include: - difficulty swallowing
- neck pain
- muscle weakness
- pain at the injection site
- muscle and bone pain
The most common side effects of XEOMIN in adults with blepharospasm include: - drooping of the eyelid
- dry eye
- vision problems
- dry mouth
The most common side effect of Xeomin in adults with upper facial lines (glabellar lines, horizontal forehead lines, and lateral canthal lines) include: - injection site bruising
These are not all the possible side effects of XEOMIN.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of XEOMIN.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or doctor for information about XEOMIN that is written for health professionals.What are the ingredients in XEOMIN?
Active ingredient: botulinum toxin type A
Inactive ingredients: human albumin and sucrose
Manufactured by: Merz Pharmaceuticals GmbH, Eckenheimer Landstrasse 100, Frankfurt Germany U.S. License Number 1830
Distributed by: Merz Pharmaceuticals, LLC, 6601 Six Forks Road, Suite 430, Raleigh, NC 27615 and Merz North America, Inc. 4133 Courtney Street, Suite 10, Franksville, WI 53126
©2024 Merz Pharmaceuticals, LLC, XEOMIN® is a registered trademark of Merz Pharma GmbH & Co KGaA.
Patent www.merztherapeutics.com/us/patents/
IN00180-02 -
Problems with swallowing, speaking, or breathing. These problems can happen hours to weeks after an injection of XEOMIN if the muscles that you use to breathe and swallow become weak after the injection. Death can happen as a complication if you have severe problems with swallowing or breathing after treatment with XEOMIN.
- PRINCIPAL DISPLAY PANEL - 50 Unit Vial Carton
- PRINCIPAL DISPLAY PANEL - 100 Unit Vial Carton
- PRINCIPAL DISPLAY PANEL - 50 Unit Vial Carton - NDC 0259-4150-01
- PRINCIPAL DISPLAY PANEL - 100 Unit Vial Carton - NDC 0259-4110-01
- PRINCIPAL DISPLAY PANEL - 200 Unit Vial Carton
-
INGREDIENTS AND APPEARANCE
XEOMIN
incobotulinumtoxina injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0259-1605 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Botulinum Toxin Type A (UNII: E211KPY694) (Botulinum Toxin Type A - UNII:E211KPY694) Botulinum Toxin Type A 50 [USP'U] Inactive Ingredients Ingredient Name Strength Albumin Human (UNII: ZIF514RVZR) Sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0259-1605-01 1 in 1 CARTON 09/01/2010 1 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125360 09/01/2010 XEOMIN
incobotulinumtoxina injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0259-1610 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Botulinum Toxin Type A (UNII: E211KPY694) (Botulinum Toxin Type A - UNII:E211KPY694) Botulinum Toxin Type A 100 [USP'U] Inactive Ingredients Ingredient Name Strength Albumin Human (UNII: ZIF514RVZR) Sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0259-1610-01 1 in 1 CARTON 09/01/2010 1 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125360 09/01/2010 XEOMIN
incobotulinumtoxina injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0259-4150 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Botulinum Toxin Type A (UNII: E211KPY694) (Botulinum Toxin Type A - UNII:E211KPY694) Botulinum Toxin Type A 50 [USP'U] Inactive Ingredients Ingredient Name Strength Albumin Human (UNII: ZIF514RVZR) Sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0259-4150-01 1 in 1 CARTON 09/01/2010 1 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125360 09/01/2010 XEOMIN
incobotulinumtoxina injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0259-4110 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Botulinum Toxin Type A (UNII: E211KPY694) (Botulinum Toxin Type A - UNII:E211KPY694) Botulinum Toxin Type A 100 [USP'U] Inactive Ingredients Ingredient Name Strength Albumin Human (UNII: ZIF514RVZR) Sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0259-4110-01 1 in 1 CARTON 09/01/2010 1 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125360 09/01/2010 XEOMIN
incobotulinumtoxina injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0259-1620 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Botulinum Toxin Type A (UNII: E211KPY694) (Botulinum Toxin Type A - UNII:E211KPY694) Botulinum Toxin Type A 200 [USP'U] Inactive Ingredients Ingredient Name Strength Albumin Human (UNII: ZIF514RVZR) Sucrose (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0259-1620-01 1 in 1 CARTON 11/20/2015 1 200 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:0259-1620-10 1 in 1 CARTON 11/20/2015 2 200 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125360 11/20/2015 Labeler - Merz Pharmaceuticals, LLC (126209282) Establishment Name Address ID/FEI Business Operations Merz Pharma GmbH & Co. KGaA 342543051 MANUFACTURE(0259-1605, 0259-1610, 0259-4150, 0259-4110, 0259-1620)