Label: VIBATIV- telavancin hydrochloride injection, powder, lyophilized, for solution


-
Contains inactivated NDC Code(s)
NDC Code(s): 52118-001-01, 52118-002-01 - Packager: Theravance, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 31, 2014
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VIBATIV® (telavancin) safely and effectively. See full prescribing information for VIBATIV.
VIBATIV® (telavancin) for injection, for intravenous use
Initial U.S. Approval: 2009WARNINGS
- Patients with pre-existing moderate/severe renal impairment (CrCl ≤50 mL/min)who were treated with VIBATIV for hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia had increased mortality observed versus vancomycin. Use of VIBATIV in patients with pre-existing moderate/severe renal impairment (CrCl ≤50 mL/min) should be considered only when the anticipated benefit to the patient outweighs the potential risk. (5.1)
- Nephrotoxicity: New onset or worsening renal impairment has occurred. Monitor renal function in all patients. (5.3)
- Women of childbearing potential should have a serum pregnancy test prior to administration of VIBATIV. (5.4, 8.1)
- Avoid use of VIBATIV during pregnancy unless potential benefit to the patient outweighs potential risk to the fetus. (8.1)
- Adverse developmental outcomes observed in 3 animal species at clinically relevant doses raise concerns about potential adverse developmental outcomes in humans. (8.1)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of VIBATIV and other antibacterial drugs VIBATIV should only be used to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1)
RECENT MAJOR CHANGES
Warnings and Precautions (6/2013)
Indications and Usage (1/2013)
Dosage and Administration (1/2013)
Adverse Reactions (6/2013)
Contraindications (6/2013)
INDICATIONS AND USAGE
VIBATIV is a lipoglycopeptide antibacterial drug indicated for the treatment of the following infections in adult patients caused by designated susceptible bacteria:
DOSAGE AND ADMINISTRATION
- Complicated skin and skin structure infections (cSSSI):
- Hospital-acquired and ventilator-associated bacterial pneumonia (HABP/VABP):
aCalculate using the Cockcroft-Gault formula and ideal body weight (IBW). Use actual body weight if < IBW. (12.3)
Creatinine Clearancea(CrCl)
(mL/min)VIBATIV Dosage Regimen >50 10 mg/kg every 24 hours 30-50 7.5 mg/kg every 24 hours 10-<30 10 mg/kg every 48 hours Insufficient data are available to make a dosing recommendation for patients with CrCl <10 mL/min, including patients on hemodialysis.
DOSAGE FORMS AND STRENGTHS
Single-use vials containing either 250 or 750 mg telavancin. (3)
WARNINGS AND PRECAUTIONS
- Decreased efficacy among patients treated for skin and skin structure infections with moderate/severe pre-existing renal impairment: Consider these data when selecting antibacterial therapy for patients with baseline CrCl ≤50 mL/min. (5.2)
- Hypersensitivity reactions: Serious and potentially fatal hypersensitivity reactions, including anaphylactic reactions, may occur after first or subsequent doses. VIBATIV should be used with caution in patients with known hypersensitivity to vancomycin. (5.5, 6.2)
- Infusion-related reactions: Administer VIBATIV over at least 60 minutes to minimize infusion-related reactions. (5.6)
- Clostridium difficile-associated disease: May range from mild diarrhea to fatal colitis. Evaluate if diarrhea occurs. (5.7)
- QTc prolongation: Avoid use in patients at risk. Use with caution in patients taking drugs known to prolong the QT interval. (5.9)
- Coagulation test interference: Telavancin interferes with some laboratory coagulation tests, including prothrombin time, international normalized ratio, and activated partial thromboplastin time. (5.10, 7.1)
ADVERSE REACTIONS
Most common adverse reaction (≥10% of patients treated with VIBATIV) in the HABP/VABP trials is diarrhea; in the cSSSI trials, the most common adverse reactions (≥10% of patients treated with VIBATIV) include: taste disturbance, nausea, vomiting, and foamy urine. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Theravance, Inc. at 1-855-MED-THRX (1-855-633-8479) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2014
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNINGS
1 INDICATIONS AND USAGE
1.1 Complicated Skin and Skin Structure Infections
1.2 HABP/VABP
2 DOSAGE AND ADMINISTRATION
2.1 Complicated Skin and Skin Structure Infections
2.2 Hospital-Acquired Bacterial Pneumonia/Ventilator-Associated Bacterial Pneumonia (HABP/VABP)
2.3 Patients with Renal Impairment
2.4 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality in Patients with HABP/VABP and Pre-existing Moderate to Severe Renal Impairment (CrCl≤50 mL/min)
5.2 Decreased Clinical Response in Patients with cSSSI and Pre-existing Moderate/Severe Renal Impairment (CrCl≤50 mL/min)
5.3 Nephrotoxicity
5.4 Pregnant Women and Women of Childbearing Potential
5.5 Hypersensitivity Reactions
5.6 Infusion-Related Reactions
5.7 Clostridium difficile-Associated Diarrhea
5.8 Development of Drug-Resistant Bacteria
5.9 QTc Prolongation
5.10 Coagulation Test Interference
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
8.7 Patients with Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL TRIALS
14.1 Complicated Skin and Skin Structure Infections
14.2 HABP/VABP
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNINGS
- Patients with pre-existing moderate/severe renal impairment (CrCl ≤ 50 mL/min) who were treated with VIBATIV for hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia had increased mortality observed versus vancomycin. Use of VIBATIV in patients with pre-existing moderate/severe renal impairment (CrCl≤ 50 mL/min) should be considered only when the anticipated benefit to the patient outweighs the potential risk [see Warnings and Precautions (5.1)].
- Nephrotoxicity: New onset or worsening renal impairment has occurred. Monitor renal function in all patients [see Warnings and Precautions (5.3)].
- Women of childbearing potential should have a serum pregnancy test prior to administration of VIBATIV [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- Avoid use of VIBATIV during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- Adverse developmental outcomes observed in 3 animal species at clinically relevant doses raise concerns about potential adverse developmental outcomes in humans [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)]
-
1 INDICATIONS AND USAGE
To reduce the development of drug-resistant bacteria and maintain the effectiveness of VIBATIV and other antibacterial drugs, VIBATIV should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Combination therapy may be clinically indicated if the documented or presumed pathogens include Gram-negative organisms.
Appropriate specimens for bacteriological examination should be obtained in order to isolate and identify the causative pathogens and to determine their susceptibility to telavancin. VIBATIV may be initiated as empiric therapy before results of these tests are known.
1.1 Complicated Skin and Skin Structure Infections
VIBATIV is indicated for the treatment of adult patients with complicated skin and skin structure infections (cSSSI) caused by susceptible isolates of the following Gram-positive microorganisms: Staphylococcus aureus (including methicillin-susceptible and -resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus), or Enterococcus faecalis (vancomycin-susceptible isolates only).
1.2 HABP/VABP
VIBATIV is indicated for the treatment of adult patients with hospital-acquired and ventilator-associated bacterial pneumonia (HABP/VABP), caused by susceptible isolates of Staphylococcus aureus (including methicillin-susceptible and -resistant isolates). VIBATIV should be reserved for use when alternative treatments are not suitable.
-
2 DOSAGE AND ADMINISTRATION
2.1 Complicated Skin and Skin Structure Infections
The recommended dosing for VIBATIV is 10 mg/kg administered over a 60-minute period in patients ≥18 years of age by intravenous infusion once every 24 hours for 7 to 14 days. The duration of therapy should be guided by the severity and site of the infection and the patient's clinical progress.
2.2 Hospital-Acquired Bacterial Pneumonia/Ventilator-Associated Bacterial Pneumonia (HABP/VABP)
The recommended dosing for VIBATIV is 10 mg/kg administered over a 60-minute period in patients ≥18 years of age by intravenous infusion once every 24 hours for 7 to 21 days. The duration of therapy should be guided by the severity of the infection and the patient's clinical progress.
2.3 Patients with Renal Impairment
Because telavancin is eliminated primarily by the kidney, a dosage adjustment is required for patients whose creatinine clearance is ≤50 mL/min, as listed in Table 1 [see Clinical Pharmacology (12.3)].
Table 1: Dosage Adjustment in Adult Patients with Renal Impairment aCalculate using the Cockcroft-Gault formula and ideal body weight (IBW). Use actual body weight if it is less than IBW. (12.3)
Creatinine Clearancea(CrCl)
(mL/min)VIBATIV Dosage Regimen >50 10 mg/kg every 24 hours 30-50 7.5 mg/kg every 24 hours 10-<30 10 mg/kg every 48 hours There is insufficient information to make specific dosage adjustment recommendations for patients with end-stage renal disease (CrCl <10 mL/min), including patients undergoing hemodialysis.
2.4 Preparation and Administration
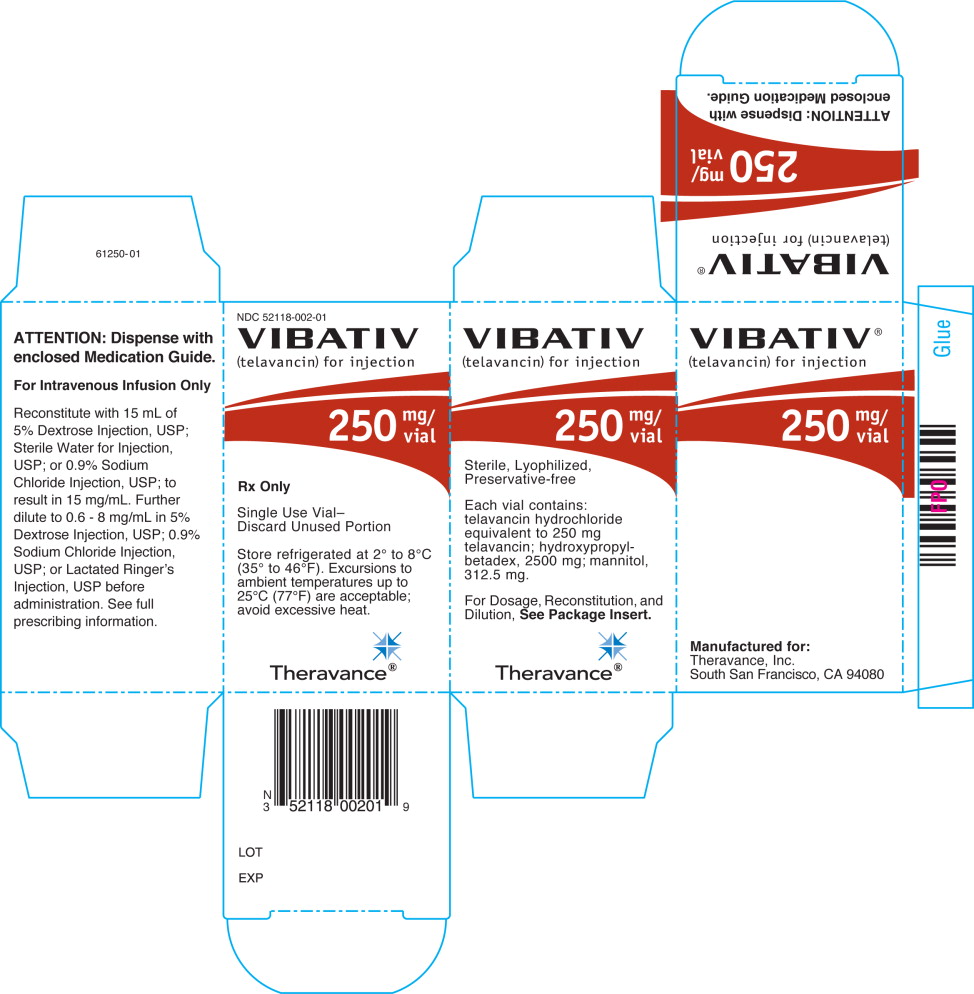
250 mg vial: Reconstitute the contents of a VIBATIV 250 mg vial with 15 mL of 5% Dextrose Injection, USP; Sterile Water for Injection, USP; or 0.9% Sodium Chloride Injection, USP. The resultant solution has a concentration of 15 mg/mL (total volume of approximately 17.0 mL).
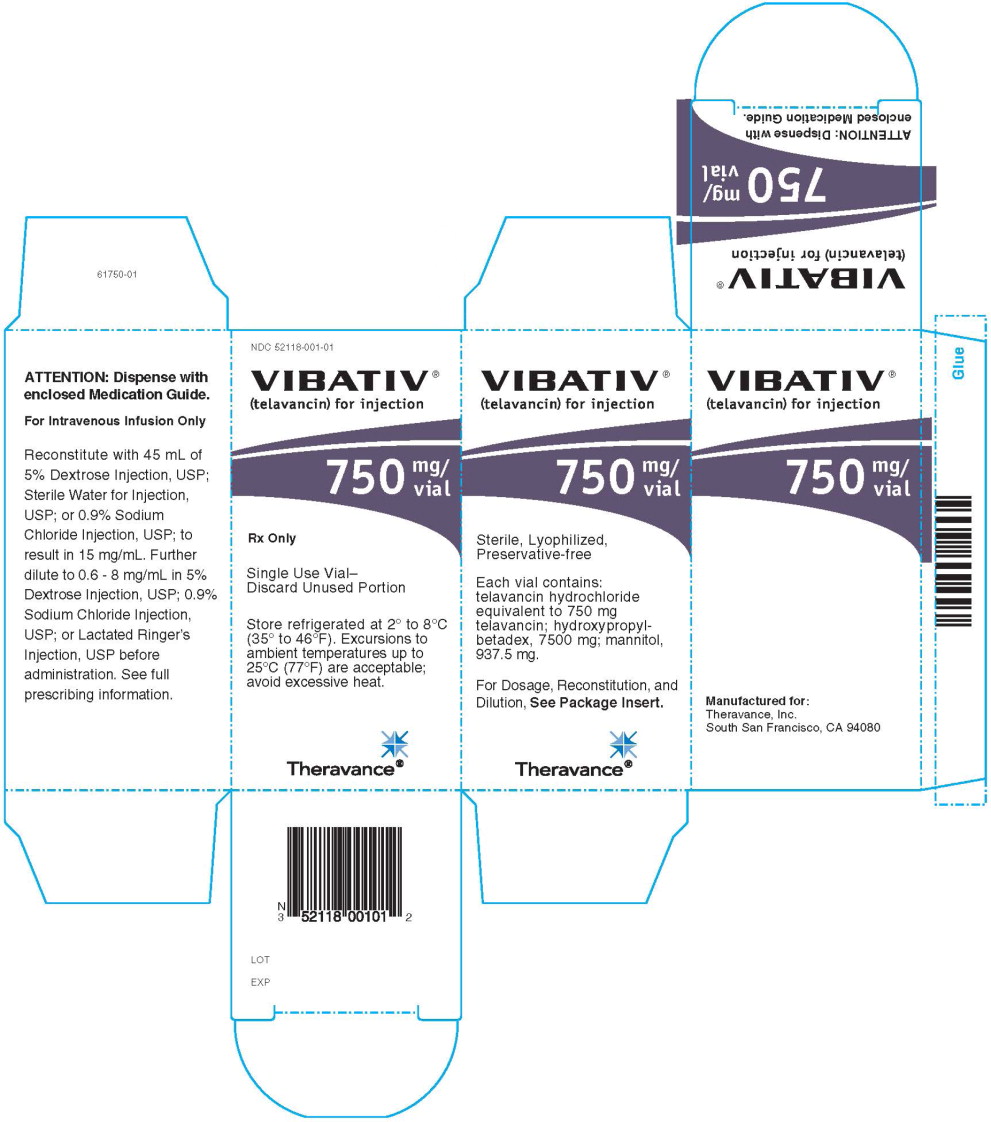
750 mg vial: Reconstitute the contents of a VIBATIV 750 mg vial with 45 mL of 5% Dextrose Injection, USP; Sterile Water for Injection, USP; or 0.9% Sodium Chloride Injection, USP. The resultant solution has a concentration of 15 mg/mL (total volume of approximately 50.0 mL).
To minimize foaming during product reconstitution, allow the vacuum of the vial to pull the diluent from the syringe into the vial. Do not forcefully inject the diluent into the vial. Do not forcefully shake the vial and do not shake final infusion solution.
The following formula can be used to calculate the volume of reconstituted VIBATIV solution required to prepare a dose:
-
Telavancin dose (mg) = 10 mg/kg or 7.5 mg/kg x patient weight (in kg) (see Table 1)
-
Volume of reconstituted solution (mL) =
Telavancin dose (mg)
15 mg/mL
For doses of 150 to 800 mg, the appropriate volume of reconstituted solution must be further diluted in 100 to 250 mL prior to infusion. Doses less than 150 mg or greater than 800 mg should be further diluted in a volume resulting in a final concentration of 0.6 to 8 mg/mL. Appropriate infusion solutions include: 5% Dextrose Injection, USP; 0.9% Sodium Chloride Injection, USP; or Lactated Ringer's Injection, USP. The dosing solution should be administered by intravenous infusion over a period of 60 minutes.
Reconstitution time is generally under 2 minutes, but can sometimes take up to 20 minutes. Mix thoroughly to reconstitute and check to see if the contents have dissolved completely. Parenteral drug products should be inspected visually for particulate matter prior to administration. Discard the vial if the vacuum did not pull the diluent into the vial.
Since no preservative or bacteriostatic agent is present in this product, aseptic technique must be used in preparing the final intravenous solution. Studies have shown that the reconstituted solution in the vial should be used within 12 hours when stored at room temperature or within 7 days under refrigeration at 2 to 8°C (36 to 46°F). The diluted (dosing) solution in the infusion bag should be used within 12 hours when stored at room temperature or used within 7 days when stored under refrigeration at 2 to 8°C (36 to 46°F). However, the total time in the vial plus the time in the infusion bag should not exceed 12 hours at room temperature and 7 days under refrigeration at 2 to 8°C (36 to 46°F). The diluted (dosing) solution in the infusion bag can also be stored at -30 to -10°C (-22 to 14°F) for up to 32 days.
VIBATIV is administered intravenously. Because only limited data are available on the compatibility of VIBATIV with other IV substances, additives or other medications should not be added to VIBATIV single-use vials or infused simultaneously through the same IV line. If the same intravenous line is used for sequential infusion of additional medications, the line should be flushed before and after infusion of VIBATIV with 5% Dextrose Injection, USP; 0.9% Sodium Chloride Injection, USP; or Lactated Ringer's Injection, USP.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality in Patients with HABP/VABP and Pre-existing Moderate to Severe Renal Impairment (CrCl≤50 mL/min)
In the analysis of patients (classified by the treatment received) in the two combined HABP/VABP trials with pre-existing moderate/severe renal impairment (CrCl≤50 mL/min), all-cause mortality within 28 days of starting treatment was 95/241 (39%) in the VIBATIV group, compared with 72/243 (30%) in the vancomycin group. All-cause mortality at 28 days in patients without pre-existing moderate/severe renal impairment (CrCl>50 mL/min) was 86/510 (17%) in the VIBATIV group and 92/510 (18%) in the vancomycin group. Therefore, VIBATIV use in patients with baseline CrCl≤50 mL/min should be considered only when the anticipated benefit to the patient outweighs the potential risk [see Adverse Reactions, Clinical Trials Experience (6.1) and Clinical Trials, HABP/VABP (14.2)].
5.2 Decreased Clinical Response in Patients with cSSSI and Pre-existing Moderate/Severe Renal Impairment (CrCl≤50 mL/min)
In a subgroup analysis of the combined cSSSI trials,clinical cure rates in the VIBATIV-treated patients were lower in patients with baseline CrCl ≤50 mL/min compared with those with CrCl>50 mL/min (Table 2). A decrease of this magnitude was not observed in vancomycin-treated patients. Consider these data when selecting antibacterial therapy for use in patients with cSSSI and with baseline moderate/severe renal impairment.
Table 2: Clinical Cure by Pre-existing Renal Impairment – Clinically Evaluable Population VIBATIV
% (n/N)Vancomycin
% (n/N)cSSSI Trials CrCl>50 mL/min 87.0% (520/598) 85.9% (524/610) CrCl ≤50 mL/min 67.4% (58/86) 82.7% (67/81) 5.3 Nephrotoxicity
In both the HABP/VABP trials and the cSSSI trials, renal adverse events were more likely to occur in patients with baseline comorbidities known to predispose patients to kidney dysfunction (pre-existing renal disease, diabetes mellitus, congestive heart failure, or hypertension). The renal adverse event rates were also higher in patients who received concomitant medications known to affect kidney function (e.g., non-steroidal anti-inflammatory drugs, ACE inhibitors, and loop diuretics).
Monitor renal function (i.e., serum creatinine, creatinine clearance) in all patients receiving VIBATIV. Values should be obtained prior to initiation of treatment, during treatment (at 48- to 72-hour intervals or more frequently, if clinically indicated), and at the end of therapy. If renal function decreases, the benefit of continuing VIBATIV versus discontinuing and initiating therapy with an alternative agent should be assessed [see Dosage and Administration (2), Adverse Reactions (6), and Clinical Pharmacology (12.3)].
In patients with renal dysfunction, accumulation of the solubilizer hydroxypropyl-beta-cyclodextrin can occur [see Patients with Renal Impairment (8.6) and Clinical Pharmacology (12.3)].
5.4 Pregnant Women and Women of Childbearing Potential
Avoid use of VIBATIV during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus. VIBATIV caused adverse developmental outcomes in 3 animal species at clinically relevant doses. This raises concern about potential adverse developmental outcomes in humans.
Women of childbearing potential should have a serum pregnancy test prior to administration of VIBATIV. If not already pregnant, women of childbearing potential should use effective contraception during VIBATIV treatment [see Use in Specific Populations (8.1)].
5.5 Hypersensitivity Reactions
Serious and sometimes fatal hypersensitivity reactions, including anaphylactic reactions, may occur after first or subsequent doses. Discontinue VIBATIV at first sign of skin rash, or any other sign of hypersensitivity. Telavancin is a semi-synthetic derivative of vancomycin; it is unknown if patients with hypersensitivity reactions to vancomycin will experience cross-reactivity to telavancin. VIBATIV should be used with caution in patients with known hypersensitivity to vancomycin [see Postmarketing Experience (6.2)].
5.6 Infusion-Related Reactions
VIBATIV is a lipoglycopeptide antibacterial agent and should be administered over a period of 60 minutes to reduce the risk of infusion-related reactions. Rapid intravenous infusions of the glycopeptide class of antimicrobial agents can cause “Red-man Syndrome”-like reactions including: flushing of the upper body, urticaria, pruritus, or rash. Stopping or slowing the infusion may result in cessation of these reactions.
5.7 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported with nearly all antibacterial agents and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hyper-toxin-producing strains of C. difficile cause increased morbidity and mortality, since these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.8 Development of Drug-Resistant Bacteria
Prescribing VIBATIV in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
As with other antibacterial drugs, use of VIBATIV may result in overgrowth of nonsusceptible organisms, including fungi. Patients should be carefully monitored during therapy. If superinfection occurs, appropriate measures should be taken.
5.9 QTc Prolongation
In a study involving healthy volunteers, doses of 7.5 and 15 mg/kg of VIBATIV prolonged the QTc interval [see Clinical Pharmacology (12.2)]. Caution is warranted when prescribing VIBATIV to patients taking drugs known to prolong the QT interval. Patients with congenital long QT syndrome, known prolongation of the QTc interval, uncompensated heart failure, or severe left ventricular hypertrophy were not included in clinical trials of VIBATIV. Use of VIBATIV should be avoided in patients with these conditions.
5.10 Coagulation Test Interference
Although telavancin does not interfere with coagulation, it interfered with certain tests used to monitor coagulation (Table 3), when conducted using samples drawn 0 to 18 hours after VIBATIV administration for patients being treated once every 24 hours. Blood samples for these coagulation tests should be collected as close as possible prior to a patient's next dose of VIBATIV. Blood samples for coagulation tests unaffected by VIBATIV may be collected at any time [see Drug Interactions (7.1)].
Table 3: Coagulation Tests Affected and Unaffected by Telavancin Affected by Telavancin Unaffected by Telavancin Prothrombin time/international normalized ratio
Activated partial thromboplastin time
Activated clotting time
Coagulation based factor X activity assayThrombin time
Whole blood (Lee-White) clotting time
Platelet aggregation study
Chromogenic anti-factor Xa assay
Functional (chromogenic) factor X activity assay
Bleeding time
D-dimer
Fibrin degradation productsNo evidence of increased bleeding risk has been observed in clinical trials with VIBATIV. Telavancin has no effect on platelet aggregation. Furthermore, no evidence of hypercoagulability has been seen, as healthy subjects receiving VIBATIV have normal levels of D-dimer and fibrin degradation products.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are also discussed elsewhere in the labeling:
- Nephrotoxicity [see Warnings and Precautions (5.3)]
- Infusion-related reactions [see Warnings and Precautions (5.6)]
- Clostridium difficile-associated diarrhea [see Warnings and Precautions (5.7)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
Complicated Skin and Skin Structure Infections
The two Phase 3 cSSSI clinical trials (Trial 1 and Trial 2) for VIBATIV included 929 adult patients treated with VIBATIV at 10 mg/kg IV once daily. The mean age of patients treated with VIBATIV was 49 years (range 18-96). There was a slight male predominance (56%) in patients treated with VIBATIV, and patients were predominantly Caucasian (78%).
In the cSSSI clinical trials, <1% (8/929) patients who received VIBATIV died and <1% (8/938) patients treated with vancomycin died. Serious adverse events were reported in 7% (69/929) of patients treated with VIBATIV and most commonly included renal, respiratory, or cardiac events. Serious adverse events were reported in 5% (43/938) of vancomycin-treated patients, and most commonly included cardiac, respiratory, or infectious events. Treatment discontinuations due to adverse events occurred in 8% (72/929) of patients treated with VIBATIV, the most common events being nausea and rash (~1% each). Treatment discontinuations due to adverse events occurred in 6% (53/938) of vancomycin-treated patients, the most common events being rash and pruritus (~1% each).
The most common adverse events occurring in ≥10% of VIBATIV-treated patients observed in the VIBATIV Phase 3 cSSSI trials were taste disturbance, nausea, vomiting, and foamy urine.
Table 4 displays the incidence of treatment-emergent adverse drug reactions reported in ≥2% of patients treated with VIBATIV possibly related to the drug.
Table 4: Incidence of Treatment-Emergent Adverse Drug Reactions Reported in ≥2% of VIBATIV or Vancomycin Patients Treated in cSSSI Trial 1 and Trial 2 VIBATIV
(N=929)Vancomycin
(N=938)* Described as a metallic or soapy taste.
Body as a Whole Rigors 4% 2% Digestive System Nausea 27% 15% Vomiting 14% 7% Diarrhea 7% 8% Metabolic and Nutritional Decreased appetite 3% 2% Nervous System Taste disturbance* 33% 7% Renal System Foamy urine 13% 3% HABP/VABP
Two randomized, double-blind Phase 3 trials (Trial 1 and Trial 2) for VIBATIV included 1,503 adult patients treated with VIBATIV at 10 mg/kg IV once daily or vancomycin at 1 g IV twice daily. The mean age of patients treated with VIBATIV was 62 years (range 18-100). In patients treated with VIBATIV, 69% of the patients were white and 65% were male. In the combined VIBATIV group, 29% were VAP and 71% were HAP patients.
Table 5 summarizes deaths using Kaplan-Meier estimates at Day 28 as stratified by baseline creatinine clearance categorized into four groups. Patients with pre-existing moderate/severe renal impairment (CrCl≤50 mL/min) who were treated with VIBATIV for HABP/VABP had increased mortality observed versus vancomycin in both the trials.
Table 5: 28-Day Mortality (Kaplan-Meier Estimates) Stratified by Baseline Creatinine Clearance — All-Treated Analysis Population CrCl (mL/min) Trial 1 Trial 2 VIBATIV
N (%)Vancomycin
N (%)Difference
(95% CI)VIBATIV
N (%)Vancomycin
N (%)Difference
(95% CI)>80 143 (12.2%) 152 (14.1%) -1.8
(-9.6, 6.0)181 (10.5%) 181 (18.7%) -8.2
(-15.5, -0.9)>50-80 88 (27.4%) 88 (17.7%) 9.7
(-2.7, 22.1)96 (25.6%) 90 (27.1%) -1.5
(-14.4, 11.3)30-50 80 (34.7%) 83 (23.1%) 11.5
(-2.5, 25.5)62 (27.7%) 68 (23.7%) 4.0
(-11.1, 19.1)<30 61 (44.3%) 51 (37.3%) 7.0
(-11.2, 25.2)38 (61.1%) 41 (42.1%) 19.0
(-2.9, 40.8)Serious adverse events were reported in 31% of patients treated with VIBATIV and 26% of patients who received vancomycin. Treatment discontinuations due to adverse events occurred in 8% (60/751) of patients who received VIBATIV, the most common events being acute renal failure and electrocardiogram QTc interval prolonged (~1% each). Treatment discontinuations due to adverse events occurred in 5% (40/752) of vancomycin-patients, the most common events being septic shock and multi-organ failure (<1%).
Table 6 displays the incidence of treatment-emergent adverse drug reactions reported in ≥ 5% of HABP/VABP patients treated with VIBATIV possibly related to the drug.
Table 6: Incidence of Treatment-Emergent Adverse Drug Reactions Reported in ≥5% of VIBATIV or Vancomycin Patients Treated in HABP/VABP Trial 1 and Trial 2 VIBATIV
(N=751)Vancomycin
(N=752)Nausea 5% 4% Vomiting 5% 4% Renal Failure Acute 5% 4% Complicated Skin and Skin Structure Infections
In cSSSI trials, the incidence of renal adverse events indicative of renal impairment (increased serum creatinine, renal impairment, renal insufficiency, and/or renal failure) was 30/929 (3%) of VIBATIV-treated patients compared with 10/938 (1%) of vancomycin-treated patients. In 17 of the 30 VIBATIV-treated patients, these adverse events had not completely resolved by the end of the trials, compared with 6 of the 10 vancomycin-treated patients. Serious adverse events indicative of renal impairment occurred in 11/929 (1%) of VIBATIV-treated patients compared with 3/938 (0.3%) of vancomycin-treated patients. Twelve patients treated with VIBATIV discontinued treatment due to adverse events indicative of renal impairment compared with 2 patients treated with vancomycin.
Increases in serum creatinine to 1.5 times baseline occurred more frequently among VIBATIV-treated patients with normal baseline serum creatinine (15%) compared with vancomycin-treated patients with normal baseline serum creatinine (7%).
Fifteen of 174 (9%) VIBATIV-treated patients ≥65 years of age had adverse events indicative of renal impairment compared with 16 of 755 patients (2%) <65 years of age [see Use in Specific Populations (8.5)].
Hospital-Acquired and Ventilator-Associated Bacterial Pneumonia
In the HABP/VABP trials, the incidence of renal adverse events (increased serum creatinine, renal impairment, renal insufficiency, and/or renal failure) was 10% for VIBATIV vs. 8% for vancomycin. Of the patients who had at least one renal adverse event, 54% in each treatment group recovered completely, recovered with sequelae, or were improving from the renal AE at the last visit. Three percent of VIBATIV-treated patients and 2% of vancomycin-treated patients experienced at least one serious renal adverse event. Renal adverse events resulted in discontinuation of study medication in 14 VIBATIV-treated patients (2%) and 7 vancomycin-treated patients (1%).
Increases in serum creatinine to 1.5 times baseline occurred more frequently among VIBATIV-treated patients (16%) compared with vancomycin-treated patients (10%).
Forty-four of 399 (11.0%) VIBATIV-treated patients ≥65 years of age had adverse events indicative of renal impairment compared with 30 of 352 patients (8%) <65 years of age [see Use in Specific Populations (8.5)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VIBATIV. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Serious hypersensitivity reactions have been reported after first or subsequent doses of VIBATIV, including anaphylactic reactions. It is unknown if patients with hypersensitivity reactions to vancomycin will experience cross-reactivity to telavancin. [see Hypersensitivity Reactions (5.5)].
-
7 DRUG INTERACTIONS
7.1 Drug-Laboratory Test Interactions
Effects of Telavancin on Coagulation Test Parameters
Telavancin binds to the artificial phospholipid surfaces added to common anticoagulation tests, thereby interfering with the ability of the coagulation complexes to assemble on the surface of the phospholipids and promote clotting in vitro. These effects appear to depend on the type of reagents used in commercially available assays. Thus, when measured shortly after completion of an infusion of VIBATIV, increases in the PT, INR, aPTT, and ACT have been observed. These effects dissipate over time, as plasma concentrations of telavancin decrease.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects: Pregnancy Category C
Pregnancy Exposure Registry
There is a pregnancy registry that monitors pregnancy outcomes in women exposed to VIBATIV during pregnancy. Physicians are encouraged to register pregnant patients, or pregnant women may enroll themselves in the VIBATIV pregnancy registry by calling 1-855-MED-THRX (1-855-633-8479).
Fetal Risk Summary
All pregnancies have a background risk of birth defects (about 3%), pregnancy loss (about 15%), or other adverse outcomes regardless of drug exposure.
There are no data on VIBATIV use in pregnant women. In 3 animal species, VIBATIV exposure during pregnancy at clinically relevant doses caused reduced fetal weights and increased rates of digit and limb malformations in offspring. These data raise concern about potential adverse developmental outcomes in humans (see Data).
Clinical Considerations
Given the lack of human data and the risks suggested by animal data, avoid using VIBATIV in pregnant women unless the benefits to the patient outweigh the potential risks to the fetus.
Data
Animal Data
In embryo-fetal development studies in rats, rabbits, and minipigs, telavancin demonstrated the potential to cause limb and skeletal malformations when given intravenously during the period of organogenesis at doses up to 150, 45, or 75 mg/kg/day, respectively. These doses resulted in exposure levels approximately 1- to 2-fold the human exposure (AUC) at the maximum clinical recommended dose. Malformations observed at <1% (but absent or at lower rates in historical or concurrent controls), included brachymelia (rats and rabbits), syndactyly (rats, minipigs), adactyly (rabbits), and polydactyly (minipigs). Additional findings in rabbits included flexed front paw and absent ulna, and in the minipigs included misshapen digits and deformed front leg. Fetal body weights were decreased in rats.
In a prenatal/perinatal development study, pregnant rats received intravenous telavancin at up to 150 mg/kg/day (approximately the same AUC as observed at the maximum clinical dose) from the start of organogenesis through lactation. Offspring showed decreases in fetal body weight and an increase in the number of stillborn pups. Brachymelia was also observed. Developmental milestones and fertility of the pups were unaffected.
8.3 Nursing Mothers
It is not known whether telavancin is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when VIBATIV is administered to a nursing woman.
8.4 Pediatric Use
The safety and effectiveness of VIBATIV in pediatric patients has not been studied.
8.5 Geriatric Use
Of the 929 patients treated with VIBATIV at a dose of 10 mg/kg once daily in clinical trials of cSSSI, 174 (19%) were ≥65 years of age and 87 (9%) were ≥75 years of age. In the cSSSI trials, lower clinical cure rates were observed in patients ≥65 years of age compared with those <65 years of age. Overall, treatment-emergent adverse events occurred with similar frequencies in patients ≥65 (75% of patients) and <65 years of age (83% of patients). Fifteen of 174 (9%) patients ≥65 years of age treated with VIBATIV had adverse events indicative of renal impairment compared with 16 of 755 (2%) patients <65 years of age [see Warnings and Precautions (5.3), Clinical Trials (14.1)].
Of the 749 HABP/VABP patients treated with VIBATIV at a dose of 10 mg/kg once daily in clinical trials of HABP/VABP, 397 (53%) were ≥65 years of age and 230 (31%) were ≥75 years of age. Treatment-emergent adverse events as well as deaths and other serious adverse events occurred more often in patients ≥65 years of age than in those <65 years of age in both treatment groups.
Telavancin is substantially excreted by the kidney, and the risk of adverse reactions may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in this age group.
The mean plasma AUC values of telavancin were similar in healthy young and elderly subjects. Dosage adjustment for elderly patients should be based on renal function [see Dosage and Administration (2), Clinical Pharmacology (12.3)].
8.6 Patients with Renal Impairment
The HABP/VABP and cSSSI trials included patients with normal renal function and patients with varying degrees of renal impairment. Patients with underlying renal dysfunction or risk factors for renal dysfunction had a higher incidence of renal adverse events [see Warnings and Precautions (5.3)].
In the HABP/VABP studies higher mortality rates were observed in the VIBATIV-treated patients with baseline CrCl≤50 mL/min. Use of VIBATIV in patients with pre-existing moderate/severe renal impairment should be considered only when the anticipated benefit to the patient outweighs the potential risk [see Warnings and Precautions (5.1)].
VIBATIV-treated patients in the cSSSI studies with baseline creatinine clearance ≤50 mL/min had lower clinical cure rates. Consider these data when selecting antibacterial therapy in patients with baseline moderate/severe renal impairment (CrCl ≤50 mL/min) [see Warnings and Precautions (5.2)].
Dosage adjustment is required in patients with ≤50 mL/min renal impairment [see Dosage and Administration (2)]. There is insufficient information to make specific dosage adjustment recommendations for patients with end-stage renal disease (CrCl <10 mL/min), including patients receiving hemodialysis [see Overdosage (10), Clinical Pharmacology (12.3)].
Hydroxypropyl-beta-cyclodextrin is excreted in urine and may accumulate in patients with renal impairment. Serum creatinine should be closely monitored and, if renal toxicity is suspected, an alternative agent should be considered [see Warnings and Precautions (5.3), Clinical Pharmacology (12.3)].
8.7 Patients with Hepatic Impairment
The HABP/VABP and cSSSI trials included patients with normal hepatic function and with hepatic impairment. No dosage adjustment is recommended in patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In the event of overdosage, VIBATIV should be discontinued and supportive care is advised with maintenance of glomerular filtration and careful monitoring of renal function. Following administration of a single dose of VIBATIV 7.5 mg/kg to subjects with end-stage renal disease, approximately 5.9% of the administered dose of telavancin was recovered in the dialysate following 4 hours of hemodialysis. However, no information is available on the use of hemodialysis to treat an overdosage [see Clinical Pharmacology (12.3)].
The clearance of telavancin by continuous venovenous hemofiltration (CVVH) was evaluated in an in vitro study [see Nonclinical Toxicology (13.2)]. Telavancin was cleared by CVVH and the clearance of telavancin increased with increasing ultrafiltration rate. However, the clearance of telavancin by CVVH has not been evaluated in a clinical study; thus, the clinical significance of this finding and use of CVVH to treat an overdosage is unknown.
-
11 DESCRIPTION
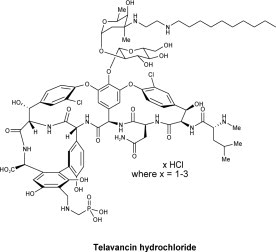
VIBATIV contains telavancin hydrochloride (Figure 1), a lipoglycopeptide antibacterial that is a synthetic derivative of vancomycin.
The chemical name of telavancin hydrochloride is vancomycin, N3''-[2-(decylamino)ethyl]-29-[[(phosphono-methyl)-amino]-methyl]- hydrochloride. Telavancin hydrochloride has the following chemical structure:
Figure 1: Telavancin Hydrochloride
Telavancin hydrochloride is an off-white to slightly colored amorphous powder with the empirical formula C80H106Cl2N11O27P•xHCl (where x = 1 to 3) and a free-base molecular weight of 1755.6. It is highly lipophilic and slightly soluble in water.
VIBATIV is a sterile, preservative-free, white to slightly colored lyophilized powder containing telavancin hydrochloride (equivalent to either 250 mg or 750 mg of telavancin as the free base) for intravenous use. The inactive ingredients are Hydroxypropylbetadex, Ph. Eur (hydroxypropyl-beta-cyclodextrin) (2500 mg per 250 mg telavancin, 7500 mg per 750 mg telavancin), mannitol (312.5 mg per 250 mg telavancin, 937.5 mg per 750 mg telavancin), and sodium hydroxide and hydrochloric acid used in minimal quantities for pH adjustment. When reconstituted, it forms a clear to slightly colored solution with a pH of 4.5 (4.0 to 5.0).
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
The antimicrobial activity of telavancin appears to best correlate with the ratio of area under the concentration-time curve to minimum inhibitory concentration (AUC/MIC) for Staphylococcus aureus based on animal models of infection. Exposure-response analyses of the clinical trials support the dose of 10 mg/kg every 24 hours.
Cardiac Electrophysiology
The effect of telavancin on cardiac repolarization was assessed in a randomized, double-blind, multiple-dose, positive-controlled, and placebo-controlled, parallel study (n=160). Healthy subjects received VIBATIV 7.5 mg/kg, VIBATIV 15 mg/kg, positive control, or placebo infused over 60 minutes once daily for 3 days. Based on interpolation of the data from VIBATIV 7.5 mg/kg and 15 mg/kg, the mean maximum baseline-corrected, placebo-corrected QTc prolongation at the end of infusion was estimated to be 12-15 msec for VIBATIV 10 mg/kg and 22 msec for the positive control (Table 7). By 1 hour after infusion the maximum QTc prolongation was 6-9 msec for VIBATIV and 15 msec for the positive control.
Table 7: Mean and Maximum QTcF Changes from Baseline Relative to Placebo QTcF1 Change from Baseline 1 Fridericia corrected
2 Upper CL from a 2-sided 90% CI on difference from placebo (msec)
Mean
(Upper 90% Confidence Limit2)
msecMaximum
(Upper 90% Confidence Limit)
msecVIBATIV 7.5 mg/kg 4.1 (7) 11.6 (16) VIBATIV 15 mg/kg 4.6 (8) 15.1 (20) Positive Control 9.5 (13) 21.6 (26) ECGs were performed prior to and during the treatment period in patients receiving VIBATIV 10 mg/kg in 3 cSSSI studies to monitor QTc intervals. In these trials, 214 of 1029 (21%) patients allocated to treatment with VIBATIV and 164 of 1033 (16%) allocated to vancomycin received concomitant medications known to prolong the QTc interval and known to be associated with definite or possible risk of torsades de pointes. The incidence of QTc prolongation >60 msec was 1.5% (15 patients) in the VIBATIV group and 0.6% (6 patients) in the vancomycin group. Nine of the 15 VIBATIV patients received concomitant medications known to prolong the QTc interval and definitely or possibly associated with a risk of torsades de pointes, compared with 1 of the 6 patients who received vancomycin. A similar number of patients in each treatment group (<1%) who did not receive a concomitant medication known to prolong the QTc interval experienced a prolongation >60 msec from baseline. In a separate analysis, 1 patient in the VIBATIV group and 2 patients in the vancomycin group experienced QTc >500 msec. No cardiac adverse events were ascribed to prolongation of the QTc interval. In the Phase 3 HABP/VABP studies, the incidence of QTc prolongation >60 msec or mean value >500 msec was 8% (52 patients) in the telavancin group and 7% (48 patients) in the vancomycin group.
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of telavancin (10 mg/kg) after a single and multiple 60-minute intravenous infusions (10 mg/kg every 24 hours) are summarized in Table 8.
Table 8: Pharmacokinetic Parameters of Telavancin in Healthy Adults, 10 mg/kg Cmax maximum plasma concentration
AUC area under concentration-time course
t1/2 terminal elimination half-life
Cl clearance
Vss apparent volume of distribution at steady state
1 Data not available
Single Dose Multiple Dose (n=42) (n=36) Cmax (mcg/mL) 93.6 ± 14.2 108 ± 26 AUC0-∞ (mcg·hr/mL) 747 ± 129 --1 AUC0-24h (mcg·hr/mL) 666 ± 107 780 125 t1/2 (hr) 8.0 ± 1.5 8.1 ± 1.5 Cl (mL/hr/kg) 13.9 ± 2.9 13.1 2.0 Vss (mL/kg) 145 ± 23 133 ± 24 In healthy young adults, the pharmacokinetics of telavancin administered intravenously were linear following single doses from 5 to 12.5 mg/kg and multiple doses from 7.5 to 15 mg/kg administered once daily for up to 7 days. Steady-state concentrations were achieved by the third daily dose.
Distribution
Telavancin binds to human plasma proteins, primarily to serum albumin, in a concentration-independent manner. The mean binding is approximately 90% and is not affected by renal or hepatic impairment.
Concentrations of telavancin in pulmonary epithelial lining fluid (ELF) and alveolar macrophages (AM) were measured through collection of bronchoalveolar lavage fluid at various times following administration of VIBATIV 10 mg/kg once daily for 3 days to healthy adults. Telavancin concentrations in ELF and AM exceeded the MIC90 for S. aureus (0.5 mcg/mL) for at least 24 hours following dosing.
Concentrations of telavancin in skin blister fluid were 40% of those in plasma (AUC0-24hr ratio) after 3 daily doses of 7.5 mg/kg VIBATIV in healthy young adults.
Metabolism
No metabolites of telavancin were detected in in vitro studies using human liver microsomes, liver slices, hepatocytes, and kidney S9 fraction. None of the following recombinant CYP 450 isoforms were shown to metabolize telavancin in human liver microsomes: CYP 1A2, 2C9, 2C19, 2D6, 3A4, 3A5, 4A11. The clearance of telavancin is not expected to be altered by inhibitors of any of these enzymes.
In a mass balance study in male subjects using radiolabeled telavancin, 3 hydroxylated metabolites were identified with the predominant metabolite (THRX-651540) accounting for <10% of the radioactivity in urine and <2% of the radioactivity in plasma. The metabolic pathway for telavancin has not been identified.
Excretion
Telavancin is primarily eliminated by the kidney. In a mass balance study, approximately 76% of the administered dose was recovered from urine and <1% of the dose was recovered from feces (collected up to 216 hours) based on total radioactivity.
Geriatric Patients
The impact of age on the pharmacokinetics of telavancin was evaluated in healthy young (range 21-42 years) and elderly (range 65-83 years) subjects. The mean CrCl of elderly subjects was 66 mL/min. Age alone did not have a clinically meaningful impact on the pharmacokinetics of telavancin [see Use in Specific Populations (8.5)].
Pediatric Patients
The pharmacokinetics of telavancin in patients less than 18 years of age have not been studied.
Gender
The impact of gender on the pharmacokinetics of telavancin was evaluated in healthy male (n=8) and female (n=8) subjects. The pharmacokinetics of telavancin were similar in males and females. No dosage adjustment is recommended based on gender.
Renal Impairment
The pharmacokinetics of telavancin were evaluated in subjects with normal renal function and subjects with varying degrees of renal impairment following administration of a single dose of telavancin 7.5 mg/kg (n=28). The mean AUC0-∞ values were approximately 13%, 29%, and 118% higher for subjects with CrCl >50 to 80 mL/min, CrCl 30 to 50 mL/min, and CrCl <30 mL/min, respectively, compared with subjects with normal renal function. Dosage adjustment is required in patients with CrCl ≤50 mL/min [see Dosage and Administration (2)].
Creatinine clearance was estimated from serum creatinine based on the Cockcroft-Gault formula:
- CrCl = [140 – age (years)] x ideal body weight (kg)* {x 0.85 for female patients}
[72 x serum creatinine (mg/dL)] -
*Use actual body weight if < ideal body weight (IBW)
IBW (male) = 50 kg + 0.9 kg/cm over 152 cm height
IBW (female) = 45.5 kg + 0.9 kg/cm over 152 cm height
Following administration of a single dose of VIBATIV 7.5 mg/kg to subjects with end-stage renal disease, approximately 5.9% of the administered dose of telavancin was recovered in the dialysate following 4 hours of hemodialysis. The effects of peritoneal dialysis have not been studied.
Following a single intravenous dose of VIBATIV 7.5 mg/kg, the clearance of hydroxypropyl-beta-cyclodextrin was reduced in subjects with renal impairment, resulting in a higher exposure to hydroxypropyl-beta-cyclodextrin. In subjects with mild, moderate, and severe renal impairment, the mean clearance values were 38%, 59%, and 82% lower, respectively, compared with subjects with normal renal function. Multiple infusions of VIBATIV may result in accumulation of hydroxypropyl-beta-cyclodextrin.
Hepatic Impairment
The pharmacokinetics of telavancin were not altered in subjects with moderate hepatic impairment (n= 8, Child-Pugh B) compared with healthy subjects with normal hepatic function matched for gender, age, and weight. The pharmacokinetics of telavancin have not been evaluated in patients with severe hepatic impairment (Child-Pugh C).
In Vitro
The inhibitory activity of telavancin against the following CYP 450 enzymes was evaluated in human liver microsomes: CYP 1A2, 2C9, 2C19, 2D6, and 3A4/5. Telavancin inhibited CYP 3A4/5 at potentially clinically relevant concentrations. Upon further evaluation in a Phase 1 clinical trial, telavancin was found not to inhibit the metabolism of midazolam, a sensitive CYP3A substrate (see below).
Midazolam
The impact of telavancin on the pharmacokinetics of midazolam (CYP 3A4/5 substrate) was evaluated in 16 healthy adult subjects following administration of a single dose of VIBATIV 10 mg/kg, intravenous midazolam 1 mg, and both. The results showed that telavancin had no impact on the pharmacokinetics of midazolam and midazolam had no effect on the pharmacokinetics of telavancin.
Aztreonam
The impact of telavancin on the pharmacokinetics of aztreonam was evaluated in 11 healthy adult subjects following administration of a single dose of VIBATIV 10 mg/kg, aztreonam 2 g, and both. Telavancin had no impact on the pharmacokinetics of aztreonam and aztreonam had no effect on the pharmacokinetics of telavancin. No dosage adjustment of telavancin or aztreonam is recommended when both drugs are coadministered.
Piperacillin-tazobactam
The impact of telavancin on the pharmacokinetics of piperacillin-tazobactam was evaluated in 12 healthy adult subjects following administration of a single dose of VIBATIV 10 mg/kg, piperacillin-tazobactam 4.5 g, and both. Telavancin had no impact on the pharmacokinetics of piperacillin-tazobactam and piperacillin-tazobactam had no effect on the pharmacokinetics of telavancin. No dosage adjustment of telavancin or piperacillin-tazobactam is recommended when both drugs are coadministered.
12.4 Microbiology
Telavancin is a semisynthetic, lipoglycopeptide antibiotic. Telavancin exerts concentration-dependent, bactericidal activity against Gram-positive organisms in vitro, as demonstrated by time-kill assays and MBC/MIC (minimum bactericidal concentration/minimum inhibitory concentration) ratios using broth dilution methodology. In vitro studies demonstrated a telavancin post-antibiotic effect ranging from 1 to 6 hours against S. aureus and other Gram-positive pathogens.
Mechanism of Action
Telavancin inhibits cell wall biosynthesis by binding to late-stage peptidoglycan precursors, including lipid II. Telavancin also binds to the bacterial membrane and disrupts membrane barrier function.
Interactions with Other Antibacterial Drugs
In vitro investigations demonstrated no antagonism between telavancin and amikacin, aztreonam, cefepime, ceftriaxone, ciprofloxacin, gentamicin, imipenem, meropenem, oxacillin, piperacillin/tazobactam, rifampin, and trimethoprim/sulfamethoxazole when tested in various combinations against telavancin-susceptible staphylococci, streptococci, and enterococci. This information is not available for other bacteria.
Cross-Resistance
Some vancomycin-resistant enterococci have a reduced susceptibility to telavancin. There is no known cross-resistance between telavancin and other classes of antibacterial drugs.
Antibacterial Activity
Telavancin has been shown to be active against most isolates of the following microorganisms both in vitro and in clinical infections as described in the Indications and Usage section [see Indications and Usage (1)]:
Facultative Gram-Positive Microorganisms
- Staphylococcus aureus (including methicillin-resistant isolates)
- Enterococcus faecalis (vancomycin-susceptible isolates only)
- Streptococcus agalactiae
- Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus)
- Streptococcus pyogenes
Greater than 90% of the following microorganisms exhibit an in vitro MIC less than or equal to the telavancin-susceptible breakpoint for organisms of similar genus shown in Table 9. The safety and effectiveness of telavancin in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials.
Facultative Gram-Positive Microorganisms
- Enterococcus faecium (vancomycin-susceptible isolates only)
- Staphylococcus haemolyticus
- Streptococcus dysgalactiae subsp. equisimilis
- Staphylococcus epidermidis
Susceptibility Test Methods
When available, the clinical microbiology laboratory should provide cumulative results of the in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas to the physician as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting an antimicrobial drug.
Dilution technique
Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MICs should be determined using a standardized procedure [see References (15)]. Standardized procedures are based on a broth dilution method or equivalent with standardized inoculum concentrations and standardized concentrations of telavancin powder. The test method treats telavancin as a water-insoluble agent. Dimethyl sulfoxide is used as solvent and diluent, and the cation-adjusted Mueller Hinton Broth test medium is supplemented with polysorbate 80 to a final concentration of 0.002%. Telavancin should not be tested by the agar dilution method. The MIC values should be interpreted according to the criteria provided in Table 9.
Diffusion technique
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure requires the use of standardized inoculum concentrations [see References (15)]. This procedure uses paper disks impregnated with 30 mcg of telavancin to test the susceptibility of microorganisms to telavancin. The disk diffusion interpretive criteria are provided in Table 9.
Table 9: Susceptibility Interpretive Criteria for Telavancin 1 The current absence of resistant isolates precludes defining any results other than “susceptible.” Isolates yielding results other than susceptible should be subjected to additional testing.
Susceptibility Interpretive Criteria1 Minimum Inhibitory Concentration (mcg/mL) Disk Diffusion Zone Diameter (mm) S I R S I R Staphylococcus aureus
(including methicillin-resistant isolates)≤ 0.12 -- -- ≥ 15 -- -- Streptococcus pyogenes
Streptococcus agalactiae≤ 0.12 -- -- ≥ 15 -- -- Streptococcus anginosus group ≤ 0.06 ≥ 15 Enterococcus faecalis (vancomycin-susceptible isolates only) ≤ 0.25 -- -- ≥ 15 -- -- A report of “susceptible" indicates that the antimicrobial is likely to inhibit growth of the pathogen if the antimicrobial compound in the blood reaches the concentrations usually achievable.
Quality Control
Standardized susceptibility test procedures require the use of laboratory control microorganisms to monitor the performance of the supplies and reagents used in the assay, and the techniques of the individuals performing the test [see References (15)]. Standard telavancin powder should provide the range of values noted in Table 10.
Quality control microorganisms are specific strains of organisms with intrinsic biological properties relating to resistance mechanisms and their genetic expression within bacteria; the specific strains used for microbiological quality control are not clinically significant.
Table 10: Acceptable Quality Control Ranges for Telavancin to be used in Validation of Susceptibility Test Results Acceptable Quality Control Ranges 1 This organism may be used for validation of susceptibility test results when testing Streptococcus spp. other than S. pneumoniae
Minimum Inhibitory Concentration (mcg/mL) Disk Diffusion Zone Diameter (mm) Enterococcus faecalis
ATCC 292120.03 – 0.12 Not applicable Staphylococcus aureus
ATCC 292130.03 - 0.12 Not applicable Staphylococcus aureus
ATCC 25923Not applicable 16-20 Streptococcus pneumoniae
ATCC 4961910.004 – 0.015 17-24 -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to determine the carcinogenic potential of telavancin have not been performed.
Neither mutagenic nor clastogenic potential of telavancin was found in a battery of tests including: assays for mutagenicity (Ames bacterial reversion), an in vitro chromosome aberration assay in human lymphocytes, and an in vivo mouse micronucleus assay.
Telavancin did not affect the fertility or reproductive performance of adult male rats (exposed to telavancin for at least 4 weeks prior to mating) or female rats (exposed to telavancin for at least 2 weeks prior to mating).
Male rats given telavancin for 6 weeks, at exposures similar to those measured in clinical studies, displayed altered sperm parameters that were reversible following an 8-week recovery period.
13.2 Animal Toxicology and/or Pharmacology
Two-week administration of telavancin in rats produced minimal renal tubular vacuolization with no changes in BUN or creatinine. These effects were not seen in studies conducted in dogs for similar duration. Four weeks of treatment resulted in reversible elevations in BUN and/or creatinine in association with renal tubular degeneration that further progressed following 13 weeks of treatment.
These effects occurred at exposures (based on AUCs) that were similar to those measured in clinical trials.
The potential effects of continuous venovenous hemofiltration (CVVH) on the clearance of telavancin were examined in an in vitro model using bovine blood. Telavancin was cleared by CVVH and the clearance of telavancin increased with increasing ultrafiltration rate [see Overdosage (10)].
-
14 CLINICAL TRIALS
14.1 Complicated Skin and Skin Structure Infections
Adult patients with clinically documented complicated skin and skin structure infections (cSSSI) were enrolled in two randomized, multinational, multicenter, double-blinded trials (Trial 1 and Trial 2) comparing VIBATIV (10 mg/kg IV every 24 hours) with vancomycin (1 g IV every 12 hours) for 7 to 14 days. Vancomycin dosages could be adjusted per site-specific practice. Patients could receive concomitant aztreonam or metronidazole for suspected Gram-negative and anaerobic infection, respectively. These trials were identical in design, enrolling approximately 69% of their patients from the United States.
The trials enrolled adult patients with cSSSI with suspected or confirmed MRSA as the primary cause of infection. The all-treated efficacy (ATe) population included all patients who received any amount of study medication according to their randomized treatment group and were evaluated for efficacy. The clinically evaluable population (CE) included patients in the ATe population with sufficient adherence to the protocol.
The ATe population consisted of 1,794 patients. Of these, 1,410 (79%) patients were clinically evaluable (CE). Patient baseline infection types were well-balanced between treatment groups and are presented in Table 11.
Table 11: Baseline Infection Types in Patients in cSSSI Trials 1 and 2 – ATe Population 1 Includes all patients randomized, treated, and evaluated for efficacy
VIBATIV
(N=884)1Vancomycin
(N=910)1Type of infection Major Abscess 375 (42.4%) 397 (43.6%) Deep/Extensive Cellulitis 309 (35.0%) 337 (37.0%) Wound Infection 139 (15.7%) 121 (13.3%) Infected Ulcer 45 (5.1%) 46 (5.1%) Infected Burn 16 (1.8%) 9 (1.0%) The primary efficacy endpoints in both trials were the clinical cure rates at a follow-up (Test of Cure) visit in the ATe and CE populations. Clinical cure rates in Trials 1 and 2 are displayed for the ATe and CE population in Table 12.
Table 12: Clinical Cure at Test-of-Cure in cSSSI Trials 1 and 2 – ATe and CE Populations 195% CI computed using a continuity correction
Trial 1 Trial 2 VIBATIV Vancomycin Difference VIBATIV Vancomycin Difference % (n/N) % (n/N) (95% CI)1 % (n/N) % (n/N) (95% CI)1 ATe 72.5% 71.6% 0.9
( -5.3, 7.2)74.7% 74.0% 0.7
( -5.1, 6.5)(309/426) (307/429) (342/458) (356/481 ) CE 84.3% 82.8% 1.5 83.9% 87.7% -3.8 (289/343) (288/348) ( -4.3, 7.3) (302/360) (315/359 ) ( -9.2, 1.5) The cure rates by pathogen for the microbiologically evaluable (ME) population are presented in Table 13.
Table 13: Clinical Cure Rates at the Test-of-Cure for the Most Common Pathogens in cSSSI Trials 1 and 2 – ME Population1 1 The ME population included patients in the CE population who had Gram-positive pathogens isolated at baseline and had central identification and susceptibility of the microbiological isolate(s).
VIBATIV
% (n/N)Vancomycin
% (n/N)Staphylococcus aureus
(MRSA)87.0%
(208/239)85.9%
(225/262)Staphylococcus aureus
(MSSA)82.0%
(132/161)85.1%
(131/154)Enterococcus faecalis 95.6%
(22/23)80.0%
(28/35)Streptococcus pyogenes 84.2%
(16/19)90.5%
(19/21)Streptococcus agalactiae 73.7%
(14/19)86.7%
(13/15)Streptococcus anginosus
group76.5%
(13/17)100.0%
(9/9)In the two cSSSI trials, clinical cure rates were similar across gender and race. Clinical cure rates in the VIBATIV clinically evaluable (CE) population were lower in patients ≥65 years of age compared with those <65 years of age. A decrease of this magnitude was not observed in the vancomycin CE population. Clinical cure rates in the VIBATIV CE population <65 years of age were 503/581 (87%) and in those ≥65 years were 88/122 (72%). In the vancomycin CE population clinical cure rates in patients <65 years of age were 492/570 (86%) and in those ≥65 years was 111/137 (82%). Clinical cure rates in the VIBATIV-treated patients were lower in patients with baseline CrCl ≤50 mL/min compared with those with CrCl >50 mL/min. A decrease of this magnitude was not observed in the vancomycin-treated patients [see Warnings and Precautions (5.2)].
14.2 HABP/VABP
Adult patients with hospital-acquired and ventilator-associated pneumonia were enrolled in two randomized, parallel-group, multinational, multicenter, double-blinded trials of identical design comparing VIBATIV (10 mg/kg IV every 24 hours) with vancomycin (1 g IV every 12 hours) for 7 to 21 days. Vancomycin dosages could be adjusted for body weight and/or renal function per local guidelines. Patients could receive concomitant aztreonam or metronidazole for suspected Gram-negative and anaerobic infection, respectively. The addition of piperacillin/tazobactam was also permitted for coverage of Gram-negative organisms if resistance to aztreonam was known or suspected. Patients with known or suspected infections due to methicillin-resistant Staphylococcus aureus were enrolled in the studies.
Of the patients enrolled across both trials, 64% were male and 70% were white.The mean age was 63 years. At baseline, more than 50% were admitted to an intensive care unit, about 23% had chronic obstructive pulmonary disease, about 29% had ventilator-associated pneumonia and about 6% had bacteremia. Demographic and baseline characteristics were generally well-balanced between treatment groups; however, there were differences between HABP/VABP Trial 1 and HABP/VABP Trial 2 with respect to a baseline history of diabetes mellitus (31% in Trial 1, 21% in Trial 2) and baseline renal insufficiency (CrCl ≤ 50 mL/min) (36% in Trial 1, 27% in Trial 2).
All-cause mortality was evaluated because there is historical evidence of treatment effect for this endpoint. This was a protocol pre-specified secondary endpoint. The 28-day all-cause mortality outcomes(overall and by baseline creatinine clearance categorization) in the group of patients who had at least one baseline Gram-positive respiratory pathogen are shown in Table 14. This group of patients included those who had mixed Gram-positive/Gram-negative infections.
Table 14: All-Cause Mortality at Day 28 in Patients with at least One Baseline Gram-Positive Pathogen aMortality rates are based on Kaplan-Meier estimates at Study Day 28. There were 84 patients (5.6%) whose survival statuses were not known up to 28 days after initiation of study drug and were considered censored at the last day known to be alive. Thirty-five of these patients were treated with VIBATIV and 45 were treated with vancomycin.
Trial 1 Trial 2 VIBATIV Vancomycin VIBATIV Vancomycin All Patients Mortalitya 28.7%
N=18724.3%
N=18024.3%
N=22422.3%
N=206Difference
(95% CI)4.4%
(-4.7%, 13.5%)2.0%
(-6.1%, 10%)CrCl ≤ 50 mL/min Mortalitya 41.8%
N=6335.4%
N=6843.9%
N=5329.6%
N=58Difference
(95% CI)6.4%
(-10.4, 23.2)14.3%
(-3.6, 32.2)CrCl > 50 mL/min Mortalitya 22.0%
N=12417.6%
N=11218.2%
N=17119.3%
N=148Difference
(95% CI)4.4%
(-5.9,14.7)-1.1%
(-9.8,7.6)The protocol-specified analysis included clinical cure rates at the TOC (7 to 14 days after the last dose of study drug) in the co-primary All-Treated (AT) and Clinically Evaluable (CE) populations (Table 15). Clinical cure was determined by resolution of signs and symptoms, no further antibacterial therapy for HABP/VABP after end-of-treatment, and improvement or no progression of baseline radiographic findings. However, the quantitative estimate of treatment effect for this endpoint has not been established.
Table 15: Clinical Response Rates in Trials 1 and 2 – AT and CE Populations aAll-Treated (AT) Population: Patients who received at least one dose of study medication
bClinically Evaluable (CE) Population: Patients who were clinically evaluable
Trial 1 Trial 2 VIBATIV Vancomycin VIBATIV Vancomycin ATa 57.5%
(214/372)59.1%
(221/374)60.2%
(227/377)60.0%
(228/380)Difference
(95% CI)-1.6%
(-8.6%, 5.5%)0.2%
(-6.8%, 7.2%)CEb 83.7%
(118/141)80.2%
(138/172)81.3%
(139/171)81.2%
(138/170)Difference
(95% CI)3.5%
(-5.1%, 12.0%)0.1%
(-8.2%, 8.4%) -
15 REFERENCES
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard - Ninth Edition. CLSI document M07-A9, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA, 2012.
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Disk Diffusion Susceptibility Tests; Approved Standard – Eleventh Edition. CLSI document M02-A11, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA, 2012.
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-fourth Informational Supplement, CLSI document M100-S24, Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA, 2014.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
- Cartons of 10 individually packaged 250 mg single-dose vials (NDC 52118-002-01)
- Cartons of 10 individually packaged 750 mg single-dose vials (NDC 52118-001-01)
Store original packages at refrigerated temperatures of 2 to 8°C (35 to 46 °F). Excursions to ambient temperatures (up to 25 °C (77 °F)) are acceptable. Avoid excessive heat.
-
17 PATIENT COUNSELING INFORMATION
See Medication Guide.
Use During Pregnancy and By Women of Childbearing Potential
Women of childbearing potential (those who have not had: complete absence of menses for at least 24 months or medically confirmed menopause, medically confirmed primary ovarian failure, a history of hysterectomy, bilateral oophorectomy, or tubal ligation) should:
- Be informed about the potential risk of fetal harm if VIBATIV is used during pregnancy
- Have a pregnancy test prior to administration of VIBATIV
- If not pregnant, use effective contraceptive methods to prevent pregnancy during VIBATIV treatment
- Notify their prescribing physician/ healthcare provider if they become pregnant during VIBATIV treatment
Pregnancy Registry
There is a pregnancy registry that monitors pregnancy outcomes in women exposed to VIBATIV during pregnancy. Physicians are encouraged to register pregnant patients, or pregnant women may enroll themselves in the pregnancy registry by calling 1-855-MED-THRX (1-855-633-8479).
Diarrhea
Diarrhea is a common problem caused by antibiotics that usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as 2 or more months after having received the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Correct Use of Antibacterial Drugs
Patients should be counseled that antibacterial drugs including VIBATIV should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When VIBATIV is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may: (1) decrease the effectiveness of immediate treatment, and (2) increase the likelihood that the bacteria will develop resistance and will not be treatable by VIBATIV or other antibacterial drugs in the future.
Common Adverse Effects
Patients should be informed about the common adverse effects of VIBATIV including diarrhea, taste disturbance, nausea, vomiting, headache, and foamy urine. Patients should be instructed to inform their healthcare provider if they develop any unusual symptom, or if any known symptom persists or worsens. Patients should be instructed to inform their healthcare provider of any other medications they are currently taking with VIBATIV, including over-the-counter medications.
Manufactured for:
Theravance, Inc.
South San Francisco, CA 94080US Patent Nos. 6,635,618 B2; 6,858,584 B2; 6,872,701 B2; 7,008,923 B2; 7,208,471 B2; 7,351,691 B2; 7,531,623 B2; 7,544,364 B2; 7,700,550 B2; 8,101,575 B2; 8,158,580 B2.
THERAVANCE®, the Theravance logo, and VIBATIV® are registered trademarks of Theravance, Inc.
-
MEDICATION GUIDE
MEDICATION GUIDE
VIBATIV® (vy-'ba-tiv)
(telavancin)
for injectionRead this Medication Guide before you receive VIBATIV. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about VIBATIV?
VIBATIV can cause serious side effects, including:
- Increased risk of death. VIBATIV was associated with an increased risk of death compared to vancomycin in people who already had kidney problems and were treated for bacterial pneumonia that you can get when you are in the hospital.
- New or worsening kidney problems. Your healthcare provider should do a blood test to check your kidneys before you start, while you receive, and after you stop receiving VIBATIV.
-
VIBATIV may harm your unborn baby. Women who can become pregnant should have a blood pregnancy test before receiving VIBATIV.
- Talk to your healthcare provider if you are pregnant or plan to become pregnant. Your healthcare provider will decide if VIBATIV is the right medicine for you.
- Women who can become pregnant should use effective birth control (contraception) while receiving VIBATIV.
- If you become pregnant while receiving VIBATIV, tell your healthcare provider right away. Talk to your healthcare provider about taking part in the VIBATIV Pregnancy Registry. This is a study to learn how VIBATIV affects pregnancy and babies. You can enroll in this registry by calling 1-855-MED-THRX (1-855-633-8479).
What is VIBATIV?
VIBATIV is a prescription antibacterial medicine used alone, or with other medicines, to treat adults with certain types of germs (bacteria) that cause:
- Serious skin infections
- Hospital-Acquired Bacterial Pneumonia (HABP)
- Ventilator-Associated Bacterial Pneumonia (VABP)
It is not known if VIBATIV is safe or effective in children under 18 years of age.
Who should not take VIBATIV?
Do not take VIBATIV if you:
- are allergic to telavancin or any of the ingredients in VIBATIV. See the end of this Medication Guide for a complete list of ingredients in VIBATIV.
What should I tell my healthcare provider before receiving VIBATIV?
Before you receive VIBATIV, tell your healthcare provider if you:
- have had a serious allergic reaction to VIBATIV or vancomycin
- have kidney problems
- have diabetes
- have or have had heart problems, including QTc prolongation or a family history of it
- have high blood pressure
- have any other medical conditions
- are breastfeeding or plan to breastfeed. It is not known if VIBATIV passes into breast milk. You and your healthcare provider should decide if you will breastfeed while receiving VIBATIV.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. VIBATIV and other medicines can affect each other causing side effects.
Especially tell your healthcare provider if you take:
- a Non-Steroidal Anti-Inflammatory Drug (NSAID)
- certain blood pressure medicines called ACE Inhibitors or ARBs
- water pills (diuretics)
- a blood thinner
- medicine to control your heart rate or rhythm (antiarrhythmics)
Ask your healthcare provider or pharmacist for a list of these medicines, if you are not sure.
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How will I receive VIBATIV?
- VIBATIV is given by your healthcare provider through a needle placed into your vein (IV infusion) slowly over 1 hour, 1 time each day, for 7 to 21 days.
- Do not stop receiving VIBATIV unless your healthcare provider tells you to, even if you feel better.
- Your healthcare provider will do blood tests before you start and while you receive VIBATIV.
What are the possible side effects of VIBATIV?
VIBATIV may cause serious side effects, including:
See “What is the most important information I should know about VIBATIV?”
-
Serious allergic reactions. Allergic reactions can happen in people who take VIBATIV, even after only one dose. Stop taking VIBATIV and get emergency medical help right away if you get any of the following symptoms of a severe allergic reaction:
- hives
- trouble breathing or swallowing
- swelling of the lips, tongue, face
- throat tightness, hoarseness
- rapid heartbeat
- faint
-
Infusion-related reactions. People who receive VIBATIV too quickly can have a certain type of skin reaction called “Red-man Syndrome”. Signs and symptoms of Red-man Syndrome can include:
- red color (flushing)
- rash
- itching
- Problems with the electrical system of your heart (QTc prolongation). Tell your healthcare provider right away if you have a change in your heartbeat such as a fast or irregular heartbeat or if you had a fainting episode.
Call your healthcare provider right away if you have any of the serious side effects listed above.
The most common side effects of VIBATIV include:
- change in your sense of taste
- nausea
- vomiting
- foamy urine
- diarrhea
Tell your healthcare provider about any side effect that bothers you or that does not go away. These are not all the possible side effects of VIBATIV. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VIBATIV?
- Store VIBATIV in the original package
- Keep VIBATIV refrigerated between 35°F to 46°F (2°C to 8°C)
- Keep out of heat
Keep VIBATIV and all medicines out of the reach of children.
General Information about the safe and effective use of VIBATIV.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use VIBATIV for a condition for which it is not prescribed.
This Medication Guide summarizes the most important information about VIBATIV. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about VIBATIV that is written for health professionals.
For more information, go to www.vibativ.com or call 1-855-MED-THRX (1-855-633-8479).
What are the ingredients in VIBATIV?
Active ingredient: telavancin hydrochloride
Inactive ingredients: hydroxypropylbetadex, Ph. Eur (hydroxypropyl-beta-cyclodextrin), mannitol, sodium hydroxide and hydrochloric acid
This Medication Guide has been approved by the U.S. Food and Drug Administration
Manufactured for:
Theravance, Inc.
South San Francisco, CA 94080
VIBATIV® is a registered trademark of Theravance, Inc.
Revised: 06/2013
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
VIBATIV
telavancin hydrochloride injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52118-001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength telavancin hydrochloride (UNII: 0701472ZG0) (telavancin - UNII:XK134822Z0) telavancin 15 mg in 1 mL Inactive Ingredients Ingredient Name Strength Hydroxypropylbetadex (0.58-0.68 MS) (UNII: 1I96OHX6EK) mannitol (UNII: 3OWL53L36A) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52118-001-01 1 in 1 CONTAINER 1 50 mL in 1 VIAL, SINGLE-USE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022110 11/05/2009 VIBATIV
telavancin hydrochloride injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52118-002 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength telavancin hydrochloride (UNII: 0701472ZG0) (telavancin - UNII:XK134822Z0) telavancin 15 mg in 1 mL Inactive Ingredients Ingredient Name Strength Hydroxypropylbetadex (0.58-0.68 MS) (UNII: 1I96OHX6EK) mannitol (UNII: 3OWL53L36A) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52118-002-01 1 in 1 CONTAINER 1 17 mL in 1 VIAL, SINGLE-USE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022110 11/05/2009 Labeler - Theravance, Inc. (036479616)