Label: INJECTAFER- ferric carboxymaltose injection injection, solution
INJECTAFER- ferric carboxymaltose injection, solution









- NDC Code(s): 0517-0602-01, 0517-0620-01, 0517-0650-01
- Packager: American Regent, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 1, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Injectafer safely and effectively. See full prescribing information for Injectafer.
INJECTAFER® (ferric carboxymaltose injection), for intravenous use
Initial U.S. Approval: 2013RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Injectafer is an iron replacement product indicated for the treatment of:
• iron deficiency anemia (IDA) in:
-
adult and pediatric patients 1 year of age and older who have either intolerance or an unsatisfactory response to oral iron. (1)
-
adult patients who have non-dialysis dependent chronic kidney disease. (1)
• iron deficiency in adult patients with heart failure and New York Heart Association class II/III to improve exercise capacity. (1)
DOSAGE AND ADMINISTRATION
- For patients weighing 50 kg or more, the recommended dosage is Injectafer 750 mg intravenously in two doses separated by at least 7 days for a total cumulative dose of 1,500 mg of iron per course. For adult patients weighing 50 kg or more, an alternative dose of Injectafer 15 mg/kg body weight up to a maximum of 1,000 mg intravenously may be administered as a single-dose per course. (2.1)
- For patients weighing less than 50 kg, the recommended dosage is Injectafer 15 mg/kg body weight intravenously in two doses separated by at least 7 days per course. (2.1)
- See Section 2.1, Table 1 for dosage in patients with iron deficiency and heart failure. (2.1)
Injectafer treatment may be repeated if IDA or iron deficiency in heart failure reoccurs. (2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 50 mg/mL (3)
- 100 mg iron/2 mL single-dose vial
- 750 mg iron/15 mL single-dose vial
- 1,000 mg iron/20 mL single-dose vial
CONTRAINDICATIONS
Hypersensitivity to Injectafer or any of its inactive components. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Observe for signs and symptoms of hypersensitivity during and after Injectafer administration for at least 30 minutes and until clinically stable following completion of each administration. (5.1)
-
Symptomatic Hypophosphatemia: Monitor serum phosphate levels in patients at risk for low serum phosphate who require a repeat course of treatment. (5.2)
- Hypertension: Monitor patients closely for signs and symptoms of hypertension following each Injectafer administration. (5.3)
ADVERSE REACTIONS
- The most common adverse reactions in adult patients (>2%) are nausea, hypertension, flushing, injection site reactions, erythema, hypophosphatemia, and dizziness. (6.1)
- The most common adverse reactions in pediatric patients (≥4%) are hypophosphatemia, injection site reactions, rash, headache, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact American Regent at 1-800-734-9236 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Pregnancy: Risk of hypersensitivity reactions which may have serious consequences for the fetus. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2023
-
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
RECENT MAJOR CHANGES
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Administration
2.3 Repeat Treatment Monitoring Safety Assessment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Symptomatic Hypophosphatemia
5.3 Hypertension
5.4 Laboratory Test Alterations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
14 CLINICAL STUDIES
14.1 Iron Deficiency Anemia
14.2 Iron Deficiency in Heart Failure
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Injectafer is indicated for the treatment of:
• iron deficiency anemia (IDA) in:
-
adult and pediatric patients 1 year of age and older who have either intolerance or an unsatisfactory response to oral iron.
-
adult patients who have non-dialysis dependent chronic kidney disease.
• iron deficiency in adult patients with heart failure and New York Heart Association class II/III to improve exercise capacity.
-
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Recommended Dosage for Treatment of Iron Deficiency Anemia
For patients weighing 50 kg or more, the recommended dosage is:
- Injectafer 750 mg intravenously in two doses separated by at least 7 days for a total cumulative dose of 1,500 mg of iron per course.
- In adult patients, Injectafer 15 mg/kg body weight up to a maximum of 1,000 mg intravenously may be administered as a single-dose per course.
For patients weighing less than 50 kg, the recommended dosage is Injectafer 15 mg/kg body weight intravenously in two doses separated by at least 7 days per course.
Recommended Dosage in Patients with Iron Deficiency with Heart Failure
See Table 1 for recommended dosage for treatment of iron deficiency in patients with heart failure and New York Heart Association class II/III to improve exercise capacity.
Table 1: Recommended Dosage in Patients with Iron Deficiency with Heart Failure
Weight less than 70 kg Weight 70 kg or more Hb (g/dL) Hb (g/dL) < 10 10 to 14
> 14 to < 15 < 10 10 to 14 > 14 to < 15
Day 1 1,000 mg 1,000 mg 500 mg 1,000 mg 1,000 mg 500 mg Week 6 500 mg No dose No dose 1,000 mg 500 mg No dose Administer a maintenance dose of 500 mg at 12, 24 and 36 weeks if serum ferritin <100 ng/mL or serum ferritin 100-300 ng/mL with transferrin saturation <20%. There are no data available to guide dosing beyond 36 weeks or with Hb ≥15 g/dL.
2.2 Preparation and Administration
Administer Injectafer intravenously, either as an undiluted slow intravenous push or by infusion. When administered via infusion, dilute up to 1,000 mg of iron in no more than 250 mL of sterile 0.9% sodium chloride injection, USP, such that the concentration of the infusion is not less than 2 mg of iron per mL and administer over at least 15 minutes.
When added to an infusion bag containing 0.9% sodium chloride injection, USP, at concentrations ranging from 2 to 4 mg of iron per mL, Injectafer solution is physically and chemically stable for 72 hours when stored at room temperature. To maintain stability, do not dilute to concentrations less than 2 mg iron/mL.
Inspect parenteral drug products visually for the absence of particulate matter and discoloration prior to administration. The product contains no preservatives. Each vial of Injectafer is intended for a single dose.
When administering Injectafer 500 or 750 mg as a slow intravenous push, give at the rate of approximately 100 mg (2 mL) per minute. For Injectafer 1,000 mg, administer as a slow intravenous push over 15 minutes. Avoid extravasation of Injectafer since brown discoloration of the extravasation site may be long lasting. Monitor for extravasation. If extravasation occurs, discontinue the Injectafer administration at that site.
Discard unused portion.
2.3 Repeat Treatment Monitoring Safety Assessment
Injectafer treatment may be repeated if IDA or iron deficiency in heart failure reoccurs. Check serum phosphate levels in patients at risk for low serum phosphate who require a repeat course of treatment or for any patient who receives a repeat course of treatment within three months [see Warnings and Precautions (5.2)]. Treat hypophosphatemia as medically indicated.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Injectafer is contraindicated in patients with a history of hypersensitivity to Injectafer or any of its components [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and fatal, have been reported in patients receiving Injectafer. Patients may present with shock, clinically significant hypotension, loss of consciousness, and/or collapse. Monitor patients for signs and symptoms of hypersensitivity during and after Injectafer administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Injectafer when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions [see Adverse Reactions (6.1, 6.2)]. In clinical trials, serious anaphylactic/anaphylactoid reactions were reported in 0.1% (2/1,775) of subjects receiving Injectafer. Other serious or severe adverse reactions potentially associated with hypersensitivity which included, but not limited to, pruritus, rash, urticaria, wheezing, or hypotension were reported in 1.5% (26/1,775) of these subjects.
5.2 Symptomatic Hypophosphatemia
Symptomatic hypophosphatemia with serious outcomes including osteomalacia and fractures requiring clinical intervention has been reported in patients treated with Injectafer in the post-marketing setting. These cases have occurred mostly after repeated exposure to Injectafer in patients with no reported history of renal impairment. However, symptomatic hypophosphatemia has been reported after one dose. Possible risk factors for hypophosphatemia include a history of gastrointestinal disorders associated with malabsorption of fat-soluble vitamins or phosphate, inflammatory bowel disease, concurrent or prior use of medications that affect proximal renal tubular function, hyperparathyroidism, vitamin D deficiency, and malnutrition. In most cases, hypophosphatemia resolved within three months.
Correct pre-existing hypophosphatemia prior to initiating therapy with Injectafer. Monitor serum phosphate levels in patients at risk for chronic low serum phosphate. Check serum phosphate levels prior to a repeat course of treatment in patients at risk for low serum phosphate and in any patient who receives a second course of therapy within three months [see Dosage and Administration (2.3)]. Treat hypophosphatemia as medically indicated.
5.3 Hypertension
In clinical studies, hypertension was reported in 4% (67/1,775) of subjects in clinical trials 1 and 2. Transient elevations in systolic blood pressure, sometimes occurring with facial flushing, dizziness, or nausea were observed in 6% (106/1,775) of subjects in these two clinical trials. These elevations generally occurred immediately after dosing and resolved within 30 minutes. Monitor patients for signs and symptoms of hypertension following each Injectafer administration [see Dosage and Administration (2)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Hypophosphatemia [see Warnings and Precautions (5.2)]
- Hypertension [see Warnings and Precautions (5.3)]
- Laboratory Test Alterations [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
Adults
In two randomized clinical studies [Studies 1 and 2, see Clinical Studies (14)], a total of 1,775 patients were exposed to Injectafer 15 mg/kg body weight up to a maximum single dose of 750 mg of iron on two occasions separated by at least 7 days up to a cumulative dose of 1,500 mg of iron.Adverse reactions reported by ≥1% of treated patients are shown in the following table.
Table 2. Adverse reactions reported in ≥1% of Study Patients in Clinical Trials 1 and 2
Injectafer
(N=1,775)
%Pooled Comparatorsa
(N=1,783)
%Oral
iron
(N=253)
%Nausea
7.2
2
1.2
Hypertension*
4
2
0.4
Flushing*
4
0.2
0
Injection site reactions*
3
3.2
0
Erythema*
3 0.6 0
Hypophosphatemia
2.1 0.1 0 Dizziness*
2.1
1.3
0.4
Vomiting
2
1
0.4
Injection Site Discoloration**
1.4
0.3
0
Headache*
1.3
1.2
0.4
Hepatic enzyme increased*
1.2
0.2
0
Dysgeusia*
1.2
2.1
0
Hypotension
1
2
0
Rash* 1 0.3 0 Constipation
0.5
0.9
3.2
a Includes oral iron and all formulations of IV iron other than Injectafer
*Grouped Terms:
Hypertension includes hypertension, blood pressure increased, and hypertensive crisis.Flushing includes flushing and hot flush.
Injection site reactions include injection site extravasation, injection site discoloration, injection site pain, injection site irritation, injection site bruising, injection site reaction, injection site discomfort, injection site erythema, injection site hematoma, injection site hemorrhage, injection site pruritus, injection site rash, and injection site swelling.
Erythema includes erythema and injection site erythema.
Dizziness includes dizziness, balance disorder, and vertigo.
**Injection site discoloration was also included in the injection site local administration reactions grouped term.
Headache includes headache and migraine.
Hepatic enzyme increased includes alanine aminotransferase increased and aspartate aminotransferase increased.
Dysgeusia includes dysgeusia and ageusia.
Rash includes rash, urticaria, skin exfoliation, blister, erythema multiforme, injection site rash, rash maculo-papular, and rash pruritic.
Other adverse reactions reported by ≥0.5% of treated patients include abdominal pain, diarrhea, gamma glutamyl transferase increased, paresthesia, and sneezing. Transient decreases in laboratory blood phosphorus levels (< 2 mg/dL) have been observed in 27% (440/1,638) of patients in clinical trials.
Pooled data from two Phase 3 studies 1VIT09030 (NCT00981045) and 1VIT09031 (NCT00982007) with a dosing regimen of Injectafer 15 mg/kg up to a maximum of 750 mg x 2 doses to a cumulative dose of 1,500 mg of iron were analyzed to compare rates of adverse reactions in two Phase 3 parallel group studies 1VIT07017 (NCT00548860) and 1VIT07018 (NCT00548691) with a dosing regimen of Injectafer 15 mg/kg up to a maximum of 1,000 mg single dose (Table 3).
Table 3. Adverse Reactions (≥1% in any Treatment Group) In Patients Receiving Two Doses of 15 mg/kg to a Maximum of 750 mg to a Cumulative Dose of 1,500 mg or a Single Dose of Injectafer 15 mg/kg to a Maximum of 1,000 mg.
Injectafer 15 mg/kg to a maximum of 750 mg x 2 doses to a cumulative dose of 1,500 mg
Injectafer 15 mg/kg to a maximum of 1,000 mg
single dose
IVIT09030 and IVIT09031b (N=1,775)
%
IVIT07017 and IVIT07018a(N=1,200)
%
Any Adverse Reaction
24
12
Injection site reactions*
3
4
Injection site extravasation**
0.2
2
Hepatic enzyme increased*
1.2
1.2
Rash*
1
1.2
Headache*
1.3
1
Dizziness*
2.1
1
Dysgeusia*
1.2
1
Nausea
7.2
1
Hypertension*
4
1
Hypophosphatemia
2.1
1
Erythema*
3
0.3
Flushing*
4
0.3
Vomiting
2
0.2
Injection site discoloration**
1.4
<0.1
Hypotension
1
<0.1
ab Included studies 1VIT07017, 1VIT07018, 1VIT09030 and 1VIT09031
*Grouped Terms
**Injection site extravasation and injection site discoloration were also included in the injection site reactions grouped term.
Pediatric Patients
The safety of Injectafer in pediatric patients was evaluated in study 1VIT17044 (NCT03523117; Study 3). Study 1VIT17044 was a randomized, active-controlled study in which 40 patients (1 to 12 years of age: 10 patients, 12 to 17 years of age: 30 patients) received Injectafer 15 mg/kg to a maximum single dose of 750 mg (whichever was smaller) on Days 0 and 7 for a maximum total dose of 1,500 mg; 38 patients evaluable for safety in the control arm received an age-dependent formulation of oral ferrous sulfate for 28 days. The median age of patients who received Injectafer was 14.5 years (range, 1-17); 83% were female; 88% White and 13% Black. The most common adverse reactions (≥4%) were hypophosphatemia, injection site reactions, rash, headache, and vomiting.
Table 4 summarizes the adverse reactions in Study 3.
Table 4. Adverse Reactions of any Grade in Pediatric Patients Receiving Injectafer in Study 3
Injectafer
(N=40)
%Oral Ferrous Sulfate
(N=38)
%Any Adverse Reactions
35 26 Hypophosphatemia* 13 0 Injection site reactions* 8 0 Rash* 8 0 Headache 5 3 Vomiting 5 3 Nasopharyngitis 3 5 Flushing 3 0 Gastrointestinal infections 3 0 Liver function test increased 3 0 Platelet count decreased 3 0 White blood cell count decreased 3 0 *Grouped Terms
Injection site reactions include infusion site hematoma, infusion site hypoesthesia and injection site pain.
Hypophosphatemia includes hypophosphatemia and blood phosphorus decreased.
Rash includes rash, exanthema and urticaria.
Patients with Iron Deficiency and Heart Failure
The safety of Injectafer was evaluated in adult patients with iron deficiency and heart failure in randomized controlled trials FAIR-HF (NCT00520780), CONFIRM-HF (NCT01453608) and AFFIRM-AHF (NCT02937454) in which 1,016 patients received Injectafer versus 857 received placebo. The overall safety profile of Injectafer was consistent across the studied indications.
6.2 Post-marketing Experience
The following adverse reactions have been identified during post approval use of Injectafer. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been reported from the post-marketing spontaneous reports with Injectafer:
- Cardiac disorders: Tachycardia
- General disorders and administration site conditions: Chest discomfort, chills, pyrexia
- Metabolism and nutrition disorders: Hypophosphatemia
- Musculoskeletal and connective tissue disorders: Arthralgia, back pain, hypophosphatemic osteomalacia
- Nervous system disorders: Syncope
- Respiratory, thoracic and mediastinal disorders: Dyspnea
- Skin and subcutaneous tissue disorders: Angioedema, erythema, pruritus, urticaria
- Pregnancy: Fetal bradycardia
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Parenteral iron administration may be associated with hypersensitivity reactions [see Warnings and Precautions (5.1)], which may have serious consequences, such as fetal bradycardia (see Clinical Considerations). Advise pregnant women of the potential risk to a fetus. Published studies and available data from postmarketing reports with intravenous Injectafer are insufficient to assess the risk of major birth defects and miscarriage.
There are risks to the mother and fetus associated with untreated IDA in pregnancy as well as risks to the fetus associated with maternal severe hypersensitivity reactions (see Clinical Considerations).In animal reproduction studies, administration of ferric carboxymaltose to rabbits during the period of organogenesis caused adverse developmental outcomes including fetal malformations and increased implantation loss at maternally toxic doses of approximately 12% to 23% of the human weekly dose of 750 mg (based on body surface area).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Untreated IDA in pregnancy is associated with adverse maternal outcomes such as post-partum anemia. Adverse pregnancy outcomes associated with IDA include increased risk for preterm delivery and low birth weight.
Fetal/Neonatal adverse reactions
Severe adverse reactions including circulatory failure (severe hypotension, shock including in the context of anaphylactic reaction) may occur in pregnant women with parenteral iron products (such as Injectafer) which may cause fetal bradycardia, especially during the second and third trimester.
Data
Human DataPublished data from randomized controlled studies, prospective observational studies and retrospective studies on the use of ferric carboxymaltose in pregnant women have not reported an association with intravenous ferric carboxymaltose and major birth defects and miscarriage. However, these studies cannot establish or exclude the absence of any drug-related risk during pregnancy.
Animal Data
Administration of ferric carboxymaltose to rats as a one-hour intravenous infusion up to 30 mg/kg/day iron on gestation days 6 to 17 did not result in adverse embryonic or fetal findings. This daily dose in rats is approximately 40% of the human weekly dose of 750 mg based on body surface area. In rabbits, ferric carboxymaltose was administered as a one-hour infusion on gestation days 6 to 19 at iron doses of 4.5, 9, 13.5, and 18 mg/kg/day. Malformations were seen starting at the daily dose of 9 mg/kg (23% of the human weekly dose of 750 mg). Spontaneous abortions occurred starting at the daily iron dose of 4.5 mg/kg (12% of the human weekly dose of 750 mg based on body surface area). Pre-implantation loss was at the highest dose. Adverse embryonic or fetal effects were observed in the presence of maternal toxicity.
A pre- and post-natal development study was conducted in rats at intravenous doses up to 18 mg/kg/day of iron (approximately 23% of the weekly human dose of 750 mg based on body surface area). There were no adverse effects on survival of offspring, their behavior, sexual maturation or reproductive parameters.
8.2 Lactation
Risk Summary
The available published data on the use of ferric carboxymaltose in lactating women demonstrate that iron is present in breast milk. Among the breastfed infants, adverse reactions included constipation and diarrhea but none of the adverse reactions reported were considered related to ferric carboxymaltose exposure through breastmilk. There is no information on the effects of ferric carboxymaltose on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Injectafer in addition to any potential adverse effects on the breastfed child from the drug or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of Injectafer for IDA in pediatric patients aged 1 year and older who have normal kidney function and have either intolerance to oral iron or have had unsatisfactory response to oral iron have been established. Use of Injectafer for this indication in this age group is supported by evidence from adequate and well-controlled studies of Injectafer in adults with additional pharmacodynamic and safety data in pediatric patients aged 1 year and older [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
Safety and effectiveness of Injectafer have not been established in pediatric patients less than 1 year of age with IDA.
Safety and effectiveness of Injectafer have not been established to improve exercise capacity in pediatric patients with ID and symptomatic heart failure.
8.5 Geriatric Use
Of the 1,775 subjects in clinical studies of Injectafer, 50% were 65 years and over, while 25% were 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Excessive dosages of Injectafer may lead to accumulation of iron in storage sites potentially leading to hemosiderosis. A patient who received Injectafer 18,000 mg over 6 months developed hemosiderosis with multiple joint disorder, walking disability, and asthenia. In the postmarketing setting, hypophosphatemic osteomalacia has been reported in patients who have received repeated high-cumulative courses of Injectafer.
-
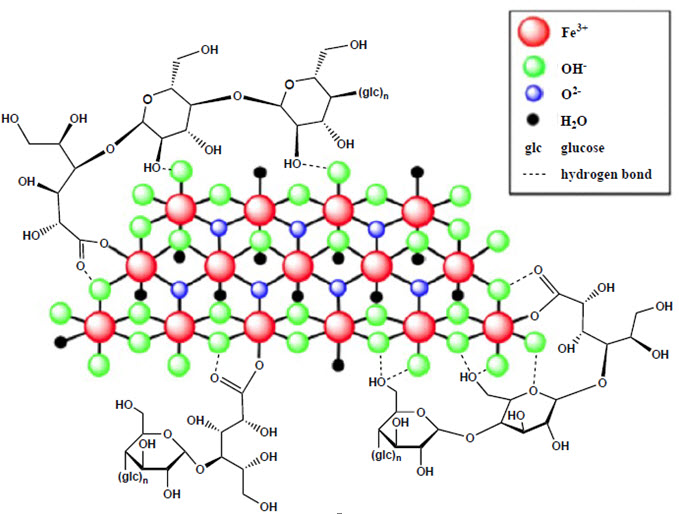
11 DESCRIPTION
Ferric carboxymaltose, an iron replacement product, is an iron carbohydrate complex with the chemical name of polynuclear iron (III)-hydroxide 4(R)-(poly-(1→4)-O-α-D-glucopyranosyl)-oxy-2(R),3(R),5(R),6-tetrahydroxy-hexanoate. It has a relative molecular weight of approximately 150,000 Da corresponding to the following empirical formula:
[FeOx(OH)y(H2O)z]n [{(C6H10O5)m (C6H12O7)}l]k,
where n ≈ 103, m ≈ 8, l ≈ 11, and k ≈ 4
(l represents the mean branching degree of the ligand).The chemical structure is presented below:
Injectafer (ferric carboxymaltose injection) is a dark brown, sterile, aqueous, isotonic colloidal solution for intravenous injection. Each mL contains 50 mg iron as ferric carboxymaltose in water for injection. Injectafer is available in 2 mL, 15 mL and 20 mL single-dose vials. Sodium hydroxide and/or hydrochloric acid may have been added to adjust the pH to 5.0-7.0.
Vial closure is not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ferric carboxymaltose is a colloidal iron (III) hydroxide in complex with carboxymaltose, a carbohydrate polymer that releases iron.
12.2 Pharmacodynamics
Using positron emission tomography (PET) it was demonstrated that red cell uptake of 59Fe and 52Fe from Injectafer ranged from 61% to 99%. In patients with iron deficiency, red cell uptake of radiolabeled iron ranged from 91% to 99% at 24 days after Injectafer dose. In patients with renal anemia, red cell uptake of radiolabeled iron ranged from 61% to 84% at 24 days after Injectafer dose.
12.3 Pharmacokinetics
After administration of a single dose of Injectafer of 100 to 1,000 mg of iron in iron deficient adult patients, maximum iron concentration of 37 µg/mL to 333 µg/mL were obtained respectively after 15 minutes to 1.21 hours post dose. The volume of distribution was estimated to be 3 L.
The iron injected or infused was rapidly cleared from the plasma, the terminal half-life ranged from 7 to 12 hours. Renal elimination of iron was negligible.
After administration of a single dose of Injectafer 15 mg/kg in pediatric patients 1-17 years of age, the maximum concentrations ranged between 124 and 418.1 μg/mL and the median time to maximum concentration was 7 minutes. The elimination half-life of Injectafer in pediatric patients was approximately 9.7 hours. The total median 72-hour exposure (AUC0-72h) after a single dose of Injectafer 15 mg/kg in pediatric patients was 4,529.7 μg∙h/mL while the median exposure after a single dose of 1,000 mg in adults was 5,875.3 μg∙h/mL. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenicity studies have not been performed with ferric carboxymaltose.
Ferric carboxymaltose was not genotoxic in the following genetic toxicology studies: in vitro microbial mutagenesis (Ames) assay, in vitro chromosome aberration test in human lymphocytes, in vitro mammalian cell mutation assay in mouse lymphoma L5178Y/TK+/- cells, in vivo mouse micronucleus test at single intravenous doses up to 500 mg/kg.
In a combined male and female fertility study, ferric carboxymaltose was administered intravenously over one hour to male and female rats at iron doses of up to 30 mg/kg. Animals were dosed 3 times per week (on Days 0, 3, and 7). There was no effect on mating function, fertility or early embryonic development. Based on body surface area, the dose of 30 mg/kg in animals is approximately 40% of the human dose of 750 mg.
-
14 CLINICAL STUDIES
14.1 Iron Deficiency Anemia
The safety and efficacy of Injectafer for treatment of IDA were evaluated in two randomized, open-label, controlled clinical trials (Trial 1 and Trial 2). In these two trials, Injectafer was administered at a dose of 15 mg/kg body weight up to a maximum single dose of 750 mg of iron on two occasions separated by at least 7 days up to a cumulative dose of 1,500 mg of iron.
Trial 1: Iron Deficiency Anemia in Patients Who Are Intolerant to Oral Iron or Have Had Unsatisfactory Response to Oral Iron
Trial 1: A Multi-center, Randomized, Active Controlled Study to Investigate the Efficacy and Safety of Intravenous Ferric Carboxymaltose (FCM) in Patients with Iron Deficiency Anemia (IDA), (NCT00982007) was a randomized, open-label, controlled clinical study in patients with IDA who had an unsatisfactory response to oral iron (Cohort 1) or who were intolerant to oral iron (Cohort 2) during the 14-day oral iron run-in period. Inclusion criteria prior to randomization included hemoglobin (Hb) <12 g/dL, ferritin ≤100 ng/mL or ferritin ≤300 ng/mL when transferrin saturation (TSAT) ≤30%. Cohort 1 subjects were randomized to Injectafer or oral iron for 14 more days. Cohort 2 subjects were randomized to Injectafer or another IV iron per standard of care [90% of subjects received iron sucrose]. The mean age of study patients was 43 years (range, 18 to 94); 94% were female; 42% were Caucasian, 32% were African American, 24% were Hispanic, and 2% were other races. The primary etiologies of IDA were heavy uterine bleeding (47%) and gastrointestinal disorders (17%).
Table 5 shows the baseline and the change in hemoglobin from baseline to highest value between baseline and Day 35 or time of intervention.
Table 5. Mean Change in Hemoglobin From Baseline to the Highest Value Between Day 35 or Time of Intervention (Modified Intent‑to‑Treat Population)
Hemoglobin (g/dL)
Mean (SD)
Cohort 1
Cohort 2
Injectafer
(N=244)Oral Iron
(N=251)Injectafer
(N=245)IV SCa
(N=237)Baseline
10.6 (1.0)
10.6 (1.0)
9.1 (1.6)
9.0 (1.5)
Highest Value
12.2 (1.1)
11.4 (1.2)
12.0 (1.2)
11.2 (1.3)
Change (from baseline to highest value)
1.6 (1.2)
0.8 (0.8)
2.9 (1.6)
2.2 (1.3)
p-value
0.001
0.001
SD=standard deviation; aIntravenous iron per standard of care
Increases from baseline in mean ferritin (264.2 ± 224.2 ng/mL in Cohort 1 and 218.2 ± 211.4 ng/mL in Cohort 2), and transferrin saturation (13 ± 16% in Cohort 1 and 20 ± 15% in Cohort 2) were observed at Day 35 in Injectafer-treated patients.
Trial 2: Iron Deficiency Anemia in Patients with Non-Dialysis Dependent Chronic Kidney Disease
Trial 2: REPAIR-IDA, Randomized Evaluation of efficacy and safety of Ferric Carboxymaltose in Patients with Iron Deficiency Anemia and Impaired Renal function, (NCT00981045) was a randomized, open-label, controlled clinical study in patients with non-dialysis dependent chronic kidney disease. Inclusion criteria included hemoglobin (Hb) ≤11.5 g/dL, ferritin ≤ 100 ng/mL or ferritin ≤300 ng/mL when transferrin saturation (TSAT) ≤ 30%. Study patients were randomized to either Injectafer or Venofer. The mean age of study patients was 67 years (range, 19 to 101); 64% were female; 54% were Caucasian, 26% were African American, 18% Hispanics, and 2% were other races.
Table 6 shows the baseline and the change in hemoglobin from baseline to highest value between baseline and Day 56 or time of intervention.
Table 6. Mean Change in Hemoglobin From Baseline to the Highest Value Between Baseline and Day 56 or Time of Intervention (Modified Intent‑to‑Treat Population)
Hemoglobin (g/dL)
Mean (SD)Injectafer
(N=1,249)Venofer
(N=1,244)Baseline
10.3 (0.8)
10.3 (0.8)
Highest Value
11.4 (1.2)
11.3 (1.1)
Change (from baseline to highest value)
1.1 (1.0)
0.9 (0.92)
Treatment Difference (95% CI)
0.21 (0.13, 0.28)
Increases from baseline in mean ferritin (734.7 ± 337.8 ng/mL) and transferrin saturation (30 ± 17%) were observed prior to Day 56 in Injectafer-treated patients.
14.2 Iron Deficiency in Heart Failure
Trial 3: FER-CARS-05 (CONFIRM-HF) was a randomized, double-blind, placebo-controlled, study in patients with iron deficiency and chronic heart failure with left ventricular ejection fraction of < 45% and New York Heart Association (NYHA) class II/III to determine whether intravenous Injectafer improves exercise capacity measured as change from baseline to 24 weeks in 6-minute walk distance (6MWD).
Iron deficiency was defined as serum ferritin <100 ng/mL or 100 to 300 ng/mL with TSAT <20%. Patients with Hb of ≥ 15 g/dl were excluded. Of the 304 patients, 150 were randomized to Injectafer and 151 to placebo. The median age of study patients was 71 years (range, 35 to 88); 46% were female; 99% were Caucasian.
At baseline, mean (SD) Hb was 12 g/dl (1.4), ferritin 57 ng/ml (45), TSAT 19 % (13.7), LVEF 37% (7), brain natriuretic peptide 770 pg/mL (973); and 57 and 43% were classified as NYHA class II and III, respectively.
At baseline, 95% of patients were treated with angiotensin converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB), 91% with beta-blocker, 59% with aldosterone antagonists, and 90% with diuretic.
The mean change in 6MWD from Baseline to Week 24 in Injectafer-treated patients was 18 meters (95% CI 4, 32), and placebo-treated patients was -7 meters (95% CI -21, 7), with between group difference of 25 meters (7, 43), p-value 0.007, favoring Injectafer. Results were generally similar within age and sex subgroups.
In Injectafer-treated patients, change from Baseline to Week 24 in serum ferritin was 269 ng/mL (229, 309), in TSAT was 9% (7, 11), and in Hb was 0.6 g/dL (0.3, 0.8).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Injectafer (ferric carboxymaltose injection) is a dark brown, non-transparent, sterile, aqueous solution.
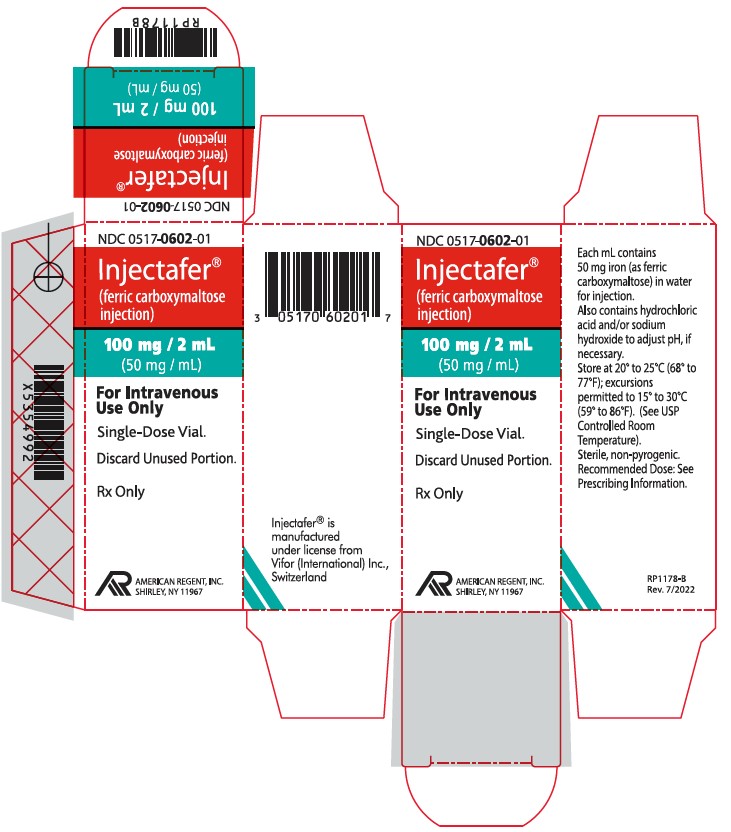
NDC 0517-0602-01 100 mg iron/2 mL Single-Dose Vial Individually Boxed
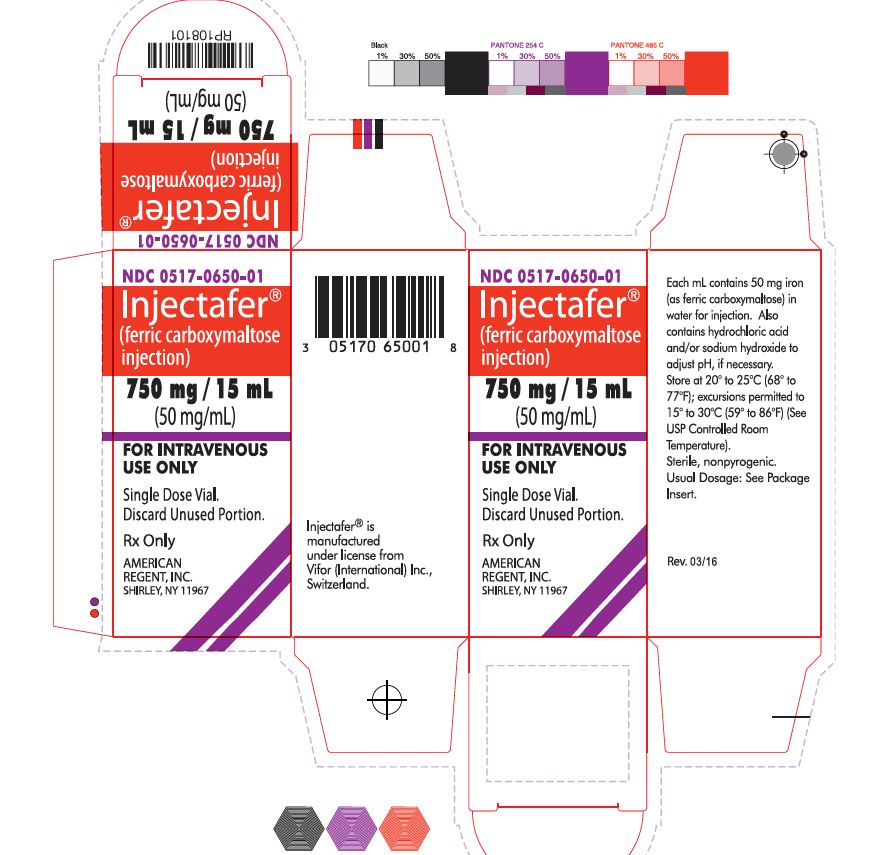
NDC 0517-0650-01 750 mg iron/15 mL Single-Dose Vial Individually Boxed
NDC 0517-0620-01 1,000 mg iron/20 mL Single-Dose Vial Individually Boxed
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). [See the USP controlled room temperature.] Do not freeze. -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information) and discuss with the patient the etiology of the iron deficiency anemia and the patient’s iron deficiency anemia treatment options.
Prior History of Reactions to Parenteral Iron Products
Question patients regarding any prior history of reactions to parenteral iron products [see Warnings and Precautions (5.1)].Serious Hypersensitivity Reactions
Advise patients to report any signs and symptoms of hypersensitivity that may develop during and following Injectafer administration, such as rash, itching, dizziness, lightheadedness, swelling, and breathing problems [see Warnings and Precautions (5.1)].
Symptomatic Hypophosphatemia
Advise patients to report any signs or symptoms of hypophosphatemia such as fatigue, muscle weakness or pain, bone and joint pain, or bone fractures [see Warnings and Precautions (5.2)].
Pregnancy
Advise pregnant women about the risk of hypersensitivity reactions which may have serious consequences for the fetus. Advise patients who may become pregnant to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].Injectafer is manufactured under license from Vifor (International) Inc, Switzerland.

RQ1052-H
-
Patient Information
INJECTAFER (in-jekt-a-fer)
(ferric carboxymaltose injection)
What is INJECTAFER?
INJECTAFER is a prescription iron replacement medicine used for the treatment of:
-
iron deficiency anemia (IDA) in:
-
adults and children 1 year of age and older who cannot tolerate iron taken by mouth (oral) or who have not responded well to oral iron.
-
adults who have chronic kidney disease who are not on dialysis (non-dialysis dependent chronic kidney disease).
-
iron deficiency in adults with mild to moderate heart failure to improve the ability to exercise (improve exercise capacity).
It is not known if INJECTAFER is safe and effective in children with IDA who are under 1 year of age.
It is not known if INJECTAFER is safe and effective in children with iron deficiency and mild to moderate heart failure to improve exercise capacity.
Do not receive INJECTAFER.
Do not receive INJECTAFER if you are allergic to ferric carboxymaltose or any of the ingredients in INJECTAFER. See the end of this Patient Information leaflet for a complete list of ingredients in INJECTAFER.
Before receiving INJECTAFER, tell your healthcare provider about all of your medical conditions, including if you:
-
have had an allergic reaction to iron given into your vein
-
have a history of trouble absorbing certain vitamins or phosphate in your body
-
have inflammatory bowel disease
-
have hyperparathyroidism
-
have low vitamin D levels
-
have high blood pressure
-
have previously received INJECTAFER
-
are pregnant or plan to become pregnant. INJECTAFER may harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with INJECTAFER.
-
are breastfeeding or plan to breastfeed. INJECTAFER passes into your breast milk. It is not known if INJECTAFER will harm your baby. Talk to your healthcare provider about the best way to feed your baby during treatment with INJECTAFER.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive INJECTAFER?
-
INJECTAFER is given into your vein (intravenously) by your healthcare provider.
-
INJECTAFER is usually given in 2 doses at least 7 days apart for IDA, or 6 weeks apart for iron deficiency with mild to moderate heart failure to improve exercise capacity.
-
If your healthcare provider decides it is right for you, INJECTAFER may be given intravenously by your healthcare provider as a single-dose treatment.
-
INJECTAFER treatment may be repeated if your healthcare provider decides it is needed.
What are the possible side effects of INJECTAFER?
INJECTAFER may cause serious side effects, including:
-
Allergic reactions. Serious life-threatening allergic reactions that can lead to death have happened in people who receive INJECTAFER and may include the following signs or symptoms:
-
low blood pressure
-
feeling dizzy or lightheaded
-
loss of consciousness
-
trouble breathing
-
swelling
-
fast heartbeat
-
cold or clammy skin
-
feet or hands turn blue
-
itching
-
rash
-
hives
-
wheezing
Your healthcare provider will watch you during and for at least 30 minutes after you receive INJECTAFER. Tell your healthcare provider right away if you develop any signs or symptoms of allergic reactions during or after treatment with INJECTAFER.
-
Symptoms of low blood phosphate levels. INJECTAFER may cause low levels of phosphate in your blood that may be serious and can lead to softening of your bones and broken bones (fractures), especially in people who have received multiple INJECTAFER treatments. Your healthcare provider may check your blood phosphate levels before a repeat treatment with INJECTAFER if you are at risk for low blood phosphate levels. If a repeat treatment is needed within 3 months of your last treatment your healthcare provider should check your blood phosphate levels.
Tell your healthcare provider if you develop any of the following signs or symptoms of low blood phosphate levels during treatment with INJECTAFER:
-
feeling very tired
-
muscle weakness or pain
-
bone or joint pain
-
broken bones
• High blood pressure. High blood pressure, sometimes with redness and warmth of the face (facial flushing), dizziness, or nausea, has happened during treatment with INJECTAFER. Your healthcare provider will check your blood pressure and check for any signs and symptoms of high blood pressure after you receive INJECTAFER.
The most common side effects of INJECTAFER in adults include:
-
nausea
-
high blood pressure
-
flushing
-
injection site reactions
-
skin redness
-
low levels of phosphate in your blood
-
dizziness
The most common side effects of INJECTAFER in children include:
-
low levels of phosphate in your blood
-
injection site reactions
-
rash
-
headache
-
vomiting
These are not all the possible side effects of INJECTAFER.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of INJECTAFER.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about INJECTAFER that is written for health professionals.
What are the ingredients in INJECTAFER?
Active ingredient: ferric carboxymaltose.
Inactive ingredients: water for injection. Sodium hydroxide or hydrochloric acid may be added to adjust pH to 5.0-7.0.

INJECTAFER is manufactured under license from Vifor (International) Inc, Switzerland.
For more information go to www.injectafer.com or call 1-800-734-9236.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 05/2023
-
- PRINCIPAL DISPLAY PANEL - 2 ML CONTAINER LABEL
- PRINCIPAL DISPLAY PANEL - 2 ML CARTON LABELING
- PRINCIPAL DISPLAY PANEL - 15 ML CONTAINER LABEL
- PRINCIPAL DISPLAY PANEL - 15 ML CARTON LABELING
- PRINCIPAL DISPLAY PANEL - 20 ML CONTAINER LABEL
- PRINCIPAL DISPLAY PANEL - 20 ML CARTON LABELING
- Serialization Label (2 mL)
- Serialization Label (15 mL)
- Serialization Label (20 mL)
-
INGREDIENTS AND APPEARANCE
INJECTAFER
ferric carboxymaltose injection injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0517-0650 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FERRIC CARBOXYMALTOSE (UNII: 6897GXD6OE) (FERRIC CATION - UNII:91O4LML611) FERRIC CATION 50 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0517-0650-01 1 in 1 BOX 08/12/2013 1 15 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203565 08/12/2013 INJECTAFER
ferric carboxymaltose injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0517-0620 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FERRIC CARBOXYMALTOSE (UNII: 6897GXD6OE) (FERRIC CATION - UNII:91O4LML611) FERRIC CATION 50 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0517-0620-01 1 in 1 BOX 06/01/2021 1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203565 06/01/2021 INJECTAFER
ferric carboxymaltose injection injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0517-0602 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FERRIC CARBOXYMALTOSE (UNII: 6897GXD6OE) (FERRIC CATION - UNII:91O4LML611) FERRIC CATION 50 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0517-0602-01 1 in 1 BOX 07/28/2022 1 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203565 06/01/2022 Labeler - American Regent, Inc. (002033710) Establishment Name Address ID/FEI Business Operations American Regent, Inc. 116981917 analysis(0517-0650, 0517-0620, 0517-0602) Establishment Name Address ID/FEI Business Operations American Regent, Inc. 606821721 manufacture(0517-0650, 0517-0620, 0517-0602) Establishment Name Address ID/FEI Business Operations Vifor (International) Inc. 482603065 api manufacture(0517-0650, 0517-0620, 0517-0602)