Label: TENIVAC (clostridium tetani toxoid antigen (formaldehyde inactivated) and corynebacterium diphtheriae toxoid antigen- formaldehyde inactivated injection, suspension




- NDC Code(s): 49281-215-10, 49281-215-15, 49281-215-58, 49281-215-88
- Packager: Sanofi Pasteur Inc.
- Category: VACCINE LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated April 12, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TENIVAC® safely and effectively. See full prescribing information for TENIVAC.
TENIVAC (Tetanus and Diphtheria Toxoids Adsorbed)
Suspension for Intramuscular Injection
Initial U.S. Approval: 2003INDICATIONS AND USAGE
TENIVAC is a vaccine indicated for active immunization for the prevention of tetanus and diphtheria in persons 7 years of age and older. (1)
DOSAGE AND ADMINISTRATION
- Each 0.5 mL dose should be administered intramuscularly. (2.5)
- Primary immunization with TENIVAC consists of 3 doses. The first 2 doses are administered 2 months apart and the third dose is administered 6-8 months after the second dose. (2.1)
- TENIVAC may be used for booster immunization against tetanus and diphtheria. Routine booster immunization against tetanus and diphtheria is recommended at 11-12 years of age and every 10 years thereafter. (2.2)
- For post-exposure diphtheria prophylaxis and for management of a tetanus prone wound, a booster dose of TENIVAC may be administered if at least 5 years have elapsed since previous receipt of a diphtheria toxoid and tetanus toxoid containing vaccine. (2.3) (2.4)
DOSAGE FORMS AND STRENGTHS
Suspension for injection supplied in 0.5 mL single-dose vials or syringes. (3)
CONTRAINDICATIONS
Severe allergic reaction (e.g., anaphylaxis) to a previous dose of TENIVAC, or any other tetanus or diphtheria toxoid-containing vaccine, or any component of this vaccine. (4.1)
WARNINGS AND PRECAUTIONS
- The tip caps of the prefilled syringes may contain natural rubber latex which may cause allergic reactions in latex sensitive individuals. (5.2)
- More frequent administration of TENIVAC than described in Dosage and Administration (2.1, 2.2, 2.3, 2.4) may be associated with increased incidence and severity of adverse reactions. (5.3)
- Persons who experienced an Arthus-type hypersensitivity reaction following a prior dose of a tetanus toxoid-containing vaccine should not receive TENIVAC more frequently than every 10 years, even for tetanus prophylaxis as part of wound management. (5.4)
- Carefully consider benefits and risks before administering TENIVAC to persons with a history of Guillain-Barré syndrome within 6 weeks of a previous tetanus toxoid-containing vaccine. (5.5)
- Syncope (fainting) has been reported following vaccination with TENIVAC. Procedures should be in place to avoid injury from fainting. (5.8)
ADVERSE REACTIONS
- The most frequent solicited injection site reaction within 0-3 days following TENIVAC was pain, reported in 78.3% of study participants 11-59 years of age and 35.3% of participants ≥60 years of age. (6.1)
- The most frequent solicited systemic reaction within 0-3 days following TENIVAC was headache, reported in 17.9% of participants, overall. (6.1)
- Other common (≥10%) solicited adverse reactions within 0-3 days following TENIVAC were injection site redness, injection site swelling, malaise, muscle weakness and pain in joints. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sanofi Pasteur Inc. at 1-800-822-2463 (1-800-VACCINE) or VAERS at 1-800-822-7967 or http://vaers.hhs.gov
DRUG INTERACTIONS
- No safety and immunogenicity data are available on the concomitant administration of TENIVAC with other US licensed vaccines. (7.1)
- If passive protection against tetanus is required, Tetanus Immune Globulin (TIG) (Human) may be administered concomitantly at a separate site with a separate needle and syringe. (7.2)
- Immunosuppressive therapies may reduce the immune response to TENIVAC. (7.3)
USE IN SPECIFIC POPULATIONS
Pre- and post-vaccination tetanus and diphtheria seroprotection rates were lower in study participants ≥65 years of age compared to younger participants. In general, rates of solicited adverse reactions were not higher in participants ≥65 years of age compared to younger participants. (8.5)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Primary Immunization
2.2 Routine Booster Immunization
2.3 Diphtheria Prophylaxis for Case Contacts
2.4 Tetanus Prophylaxis in Wound Management
2.5 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
5.2 Latex
5.3 Frequency of Administration
5.4 Arthus Reactions
5.5 Guillain-Barré Syndrome and Brachial Neuritis
5.6 Limitations of Vaccine Effectiveness
5.7 Altered Immunocompetence
5.8 Syncope
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Vaccine Administration
7.2 Tetanus Immune Globulin (Human)
7.3 Immunosuppressive Treatments
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Primary Immunization
14.2 Booster Immunization
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Primary Immunization
In persons who have not been immunized previously against tetanus and diphtheria, primary immunization with TENIVAC consists of three 0.5 mL doses. The first 2 doses are administered 2 months apart and the third dose is administered 6-8 months after the second dose.
TENIVAC may be used to complete the primary immunization series for tetanus and diphtheria, following one or two doses of Diphtheria and Tetanus Toxoids and Pertussis Vaccine Adsorbed (whole-cell DTP), Diphtheria and Tetanus Toxoids and Acellular Pertussis Vaccine Adsorbed (DTaP), and/or Diphtheria and Tetanus Toxoids Adsorbed (DT). However, the safety and efficacy of TENIVAC in such regimens have not been evaluated.
2.2 Routine Booster Immunization
TENIVAC may be used for routine booster immunization against tetanus and diphtheria in persons 7 years of age and older. Routine booster immunization against tetanus and diphtheria is recommended in children 11-12 years of age and every 10 years thereafter.
2.3 Diphtheria Prophylaxis for Case Contacts
TENIVAC may be used for post-exposure diphtheria prophylaxis in persons 7 years of age and older who have not completed primary vaccination, whose vaccination status is unknown, or who have not been vaccinated with diphtheria toxoid within the previous 5 years. Consult recommendations of the Advisory Committee on Immunization Practices for additional interventions for diphtheria prophylaxis in close contacts of diphtheria patients. (1)
2.4 Tetanus Prophylaxis in Wound Management
For active tetanus immunization in wound management of patients 7 years of age and older, a preparation containing tetanus and diphtheria toxoids is preferred instead of single-antigen tetanus toxoid to enhance diphtheria protection. (1) TENIVAC is approved for wound management of patients 7 years of age and older.
The need for active immunization with a tetanus toxoid-containing preparation, with or without passive immunization with Tetanus Immune Globulin (TIG) (Human) depends on both the condition of the wound and the patient's vaccination history. (See Table 1.)
When indicated, TIG (Human) should be administered at a separate site, with a separate needle and syringe, according to the manufacturer's package insert. If a contraindication to using tetanus toxoid-containing preparations exists in a person who has not completed a primary immunizing course of tetanus toxoid and other than a clean, minor wound is sustained, only passive immunization with TIG (Human) should be given. (1)
Table 1: Guide for use of Tetanus and Diphtheria Toxoids Adsorbed (Td) for Tetanus Prophylaxis in Routine Wound Management in Persons 7 Years of Age and Older History of Adsorbed Tetanus Toxoid (Doses) Clean, Minor Wounds All Other Wounds* Td TIG Td TIG - *
- Such as, but not limited to, wounds contaminated with dirt, puncture wounds and traumatic wounds.
- †
- If only three doses of fluid tetanus toxoid have been received, then a fourth dose of toxoid, preferably an adsorbed toxoid should be given.
- ‡
- Yes, if >10 years since last dose.
- §
- Yes, if >5 years since last dose. (More frequent boosters are not needed and can accentuate side effects.)
Unknown or < three Yes No Yes Yes ≥ Three† No‡ No No§ No 2.5 Administration
Just before use, shake the single-dose vial or syringe well until a uniform, white, cloudy suspension results. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If these conditions exist, the product should not be administered.
Administer the 0.5 mL dose of TENIVAC intramuscularly. Discard unused portion.
The preferred site is the deltoid muscle. The vaccine should not be injected into the gluteal area or areas where there may be a major nerve trunk.
Do not administer this product intravenously or subcutaneously.
TENIVAC should not be combined through reconstitution or mixed with any other vaccine.
-
3 DOSAGE FORMS AND STRENGTHS
TENIVAC is a suspension for injection available in 0.5 mL single-dose vials or syringes. [See Description (11).]
-
4 CONTRAINDICATIONS
4.1 Hypersensitivity
A severe allergic reaction (e.g., anaphylaxis) after a previous dose of TENIVAC or any other tetanus toxoid or diphtheria toxoid-containing vaccine or any other component of this vaccine is a contraindication to administration of TENIVAC. [See Description (11).] Because of uncertainty as to which component of the vaccine may be responsible, none of the components should be administered. Alternatively, such individuals may be referred to an allergist for evaluation if further immunizations are to be considered.
-
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
Epinephrine hydrochloride solution (1:1,000) and other appropriate agents and equipment must be available for immediate use in case an anaphylactic or acute hypersensitivity reaction occurs.
5.2 Latex
The tip caps of the TENIVAC prefilled syringes may contain natural rubber latex, which may cause allergic reactions in latex sensitive individuals.
5.3 Frequency of Administration
More frequent doses of TENIVAC than described in Section 2, Dosage and Administration, may be associated with increased incidence and severity of adverse reactions. [See Dosage and Administration (2.1, 2.2, 2.3, 2.4).]
5.4 Arthus Reactions
Persons who experienced an Arthus-type hypersensitivity reaction following a prior dose of a tetanus toxoid-containing vaccine usually have high serum tetanus antitoxin levels and should not receive TENIVAC more frequently than every 10 years, even for tetanus prophylaxis as part of wound management.
5.5 Guillain-Barré Syndrome and Brachial Neuritis
A review by the Institute of Medicine found evidence for a causal relation between tetanus toxoid and both brachial neuritis and Guillain-Barré syndrome. (2) If Guillain-Barré syndrome occurred within 6 weeks of receipt of prior vaccine containing tetanus toxoid, the decision to give TENIVAC or any vaccine containing tetanus toxoid should be based on careful consideration of the potential benefits and possible risks.
5.7 Altered Immunocompetence
If TENIVAC is administered to immunocompromised persons, including persons receiving immunosuppressive therapy, the expected immune response may not be obtained. [See Drug Interactions (7.3).]
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to vaccine use and for approximating rates of those events.
In a primary immunization study conducted in Canada, 18 participants, 8 of whom were 6 to 9 years of age and 10 of whom were 17 to 56 years of age, received three doses of TENIVAC. In four booster immunization studies conducted in either the US or Canada, TENIVAC was administered to 3,723 participants overall, ranging in age from 11 to 93 years.
In one of these studies, a US multi-center booster immunization study (TDC01), 2,250 adolescents and adults ages 11-59 years of age received TENIVAC in an open-label design and adults 60 years of age and over were randomized to receive either TENIVAC (N = 700) or DECAVAC (Td manufactured by Sanofi Pasteur Inc.) (N = 701). Vaccine assignment for participants ≥60 years of age was unblinded to pharmacists and vaccination nurses, but was blinded to other study personnel and participants. Among participants who received TENIVAC, overall, 80.4% were Caucasian, 3.3% Black, 5.1% Hispanic, 4.5% Asian and 6.6% other races. Among participants ≥60 years of age, the racial distribution was similar for the TENIVAC and DECAVAC groups. Among participants who received TENIVAC, the proportion of participants who were female varied by age group (44.4% of participants 11-18 years of age, 70.1% of participants 19-59 years of age and 62.4% of participants ≥60 years of age). Among participants ≥60 years of age who received DECAVAC, 57.6% were female. Nearly all (99.8%) enrolled participants and all participants in the per-protocol immunogenicity population had a reported or documented history of previous immunization against tetanus and diphtheria and, by report, had not received a vaccine containing tetanus or diphtheria toxoid within 5 years prior to enrollment.
In the US multi-center booster immunization study, solicited injection site reactions and systemic adverse events were monitored on diary cards for a subset of participants 11-59 years of age and for all participants ≥60 years of age. The incidence and severity of solicited injection site reactions and selected solicited systemic adverse events that occurred within 3 days following vaccination are shown in Table 2.
Table 2: Frequency and Severity of Selected Solicited Adverse Events Within 0–3 Days Following TENIVAC or DECAVAC in a US Study TENIVAC DECAVAC Adolescents
11 to 18 years
N = 491–492
%Adults
19 to 59 years
N = 247
%Adults
≥60 years
N = 688–695
%Adults
≥60 years
N = 686–693
%Injection Site Adverse Reactions Pain Any 80.1 74.9 35.3 29.4 Moderate* 15.0 18.2 2.9 2.3 Severe† 0.2 0.4 0.6 0.7 Redness Any 25.6 15.8 18.1 18.0 ≥35 mm to <50 mm 1.2 2.4 0.7 1.3 ≥50 mm 0.4 0.4 2.3 1.9 Swelling Any 15.0 17.0 12.1 13.0 ≥35 mm to <50 mm 1.2 2.8 1.0 1.3 ≥50 mm 1.8 2.8 1.7 1.3 Systemic Adverse Events Fever ≥37.5°C 4.3 5.7 2.5 3.8 ≥38.0°C to <39°C 0.8 1.6 0.6 0.9 ≥39°C 0.0 0.0 0.1 0.1 Headache Any 23.0 25.1 11.7 10.8 Moderate* 4.3 7.3 1.6 1.4 Severe† 0.6 0.8 0.0 0.3 Muscle Weakness Any 32.3 17.4 4.9 5.9 Moderate* 7.3 3.2 1.3 1.0 Severe† 0.6 0.4 0.1 0.1 Malaise Any 14.5 17.0 8.9 8.8 Moderate* 3.5 3.2 2.4 1.2 Severe† 0.8 0.4 0.1 0.4 Pain in Joints Any 15.7 10.9 8.5 7.4 Moderate* 2.8 1.6 2.2 1.4 Severe† 0.6 0.4 0.1 0.0 In the US booster immunization study, among participants ≥60 years of age, 7 (1.0%) participants in the TENIVAC group and 10 (1.4%) participants in the DECAVAC group experienced a serious adverse event within 30 days following vaccination. During this period, 2 (0.3%) participants 19-59 years of age and no participants 11-18 years of age experienced a serious adverse event following TENIVAC. Serious adverse events within 30 days following TENIVAC included localized infection, asthma, colonic polyp, cellulitis, angina pectoris, hip and wrist fracture, cholecystitis, chest pain and cerebrovascular accident.
There were five deaths reported during the study. All of the reported deaths were in participants ≥60 years of age and occurred >30 days post-vaccination: three in the TENIVAC group (cardiopulmonary arrest; myocardial infarction and septic shock; and unknown cause) and two in the DECAVAC group (myocardial infarction and congestive heart failure; and liver cancer).
In the primary immunization study (N = 18) in which serious adverse events were monitored for 3 days following each vaccination and in three other booster immunization studies in which serious adverse events were monitored for either four days (N = 347) or one month (N = 426) following vaccination, no serious adverse events were reported.
6.2 Postmarketing Experience
The following adverse events have been spontaneously reported during the postmarketing use of TENIVAC. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to vaccine exposure.
The following adverse events were included based on severity, frequency of reporting or the strength of causal association to TENIVAC:
-
Blood and lymphatic system disorders
Lymphadenopathy -
Immune system disorders
Allergic reactions (such as erythematous rash, maculopapular rash, urticaria and pruritus);
anaphylactic reaction (bronchospasm and angioedema). -
Nervous system disorders
Paresthesia, dizziness, syncope
Guillain-Barré syndrome -
Gastrointestinal disorders
Vomiting -
Musculoskeletal, connective tissue and bone disorders
Myalgia, pain in extremities -
General disorders and administration site conditions
Injection site reactions (including inflammation, mass, edema, induration, warmth, pruritus, cellulitis, discomfort)
Fatigue, edema peripheral
-
Blood and lymphatic system disorders
-
7 DRUG INTERACTIONS
7.1 Concomitant Vaccine Administration
No safety and immunogenicity data are available on the concomitant administration of TENIVAC with other US licensed vaccines.
7.2 Tetanus Immune Globulin (Human)
If passive protection against tetanus is required, TIG (Human) may be administered according to its prescribing information, concomitantly with TENIVAC at a separate site with a separate needle and syringe. [See Dosage and Administration (2.4).]
7.3 Immunosuppressive Treatments
Immunosuppressive therapies, including irradiation, antimetabolites, alkylating agents, cytotoxic drugs and corticosteroids (used in greater than physiologic doses), may reduce the immune response to TENIVAC. [See Warnings and Precautions (5.7).]
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. There are no adequate and well-controlled studies of TENIVAC administration in pregnant women in the U.S. There are insufficient human data from TENIVAC administered during pregnancy to establish the presence or absence of a vaccine-associated risk.
A developmental toxicity study has been performed in female rabbits administered a single human dose of TENIVAC prior to mating and during gestation. This study revealed no evidence of harm to the fetus due to TENIVAC. (See Animal data)
Data
Animal data
In a developmental toxicity study, female rabbits received a single human dose (0.5 mL) of TENIVAC by intramuscular injection 17 and 10 days prior to mating, and on gestation days 6 and 29. No adverse effects on pre-weaning development up to post-natal day 35 were observed. There were no vaccine-related fetal malformations or variations observed.
8.2 Lactation
It is not known whether TENIVAC components are excreted in human milk. Data are not available to assess the effect of administration of TENIVAC on breastfed infants or on milk production/excretion.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TENIVAC and any potential adverse effects on the breastfed child from TENIVAC or from the underlying maternal condition. For preventive vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
TENIVAC is not approved for use in infants and children younger than 7 years of age. Safety and effectiveness of TENIVAC in this age group have not been established.
8.5 Geriatric Use
In one clinical study, (TDC01) 449 participants 65 years of age and over, including 192 participants who were 75 years of age and over received a dose of TENIVAC. A lower proportion of participants 65 years of age and over had a pre-vaccination seroprotective level of antibody to tetanus toxoid and diphtheria toxin compared to adolescents and adults less than 65 years of age. The proportion of participants 65 years of age and over with a seroprotective level of antibody following TENIVAC was marginally lower for tetanus and lower for diphtheria compared to younger participants. In general, rates of solicited adverse events were not higher in participants 65 years of age and over compared to younger participants. [See Adverse Reactions (6), Clinical Pharmacology (12.1), and Clinical Studies (14.2).]
-
11 DESCRIPTION
TENIVAC, Tetanus and Diphtheria Toxoids Adsorbed, is a sterile isotonic suspension of tetanus and diphtheria toxoids adsorbed on aluminum phosphate.
Each 0.5 mL dose of TENIVAC contains the following active ingredients:
Tetanus Toxoid 5 Lf Diphtheria Toxoid 2 Lf Other ingredients per 0.5 mL dose include 1.5 mg of aluminum phosphate (0.33 mg of aluminum) as the adjuvant and ≤5.0 mcg of residual formaldehyde.
Clostridium tetani is grown in modified Mueller-Miller casamino acid medium without beef heart infusion. (3) Tetanus toxin is detoxified with formaldehyde and purified by ammonium sulfate fractionation and diafiltration. Corynebacterium diphtheriae is grown in modified Mueller's growth medium. (4) After purification by ammonium sulfate fractionation, diphtheria toxin is detoxified with formaldehyde and diafiltered. Tetanus and diphtheria toxoids are individually adsorbed onto aluminum phosphate.
The adsorbed tetanus and diphtheria toxoids are combined with aluminum phosphate (as adjuvant), sodium chloride and water for injection. This product contains no preservative.
In the guinea pig potency test, the tetanus toxoid component induces at least 2 neutralizing units/mL of serum and the diphtheria toxoid component induces at least 0.5 neutralizing units/mL of serum.
The tip caps of the prefilled syringes may contain natural rubber latex. The vial stoppers do not contain latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tetanus
Tetanus is an acute disease caused by an extremely potent neurotoxin produced by C tetani. Protection against disease is due to the development of neutralizing antibodies to tetanus toxin. A serum tetanus antitoxin level of at least 0.01 IU/mL, measured by neutralization assay is considered the minimum protective level. (5) (6) A tetanus antitoxoid level of ≥0.1 IU/mL as measured by the ELISA used in some clinical studies of TENIVAC is considered protective.
Diphtheria
Diphtheria is an acute toxin-mediated disease caused by toxigenic strains of C diphtheriae. Protection against disease is due to the development of neutralizing antibodies to diphtheria toxin. A serum diphtheria antitoxin level of 0.01 IU/mL is the lowest level giving some degree of protection. Antitoxin levels of at least 0.1 IU/mL are generally regarded as protective. (5) A level of at least of 1.0 IU/mL has been associated with long-term protection. (7)
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
TENIVAC has not been evaluated for carcinogenic or mutagenic potential or impairment of male fertility in animals. Vaccination of female rabbits with TENIVAC had no effects on fertility. [See Use in Specific Populations (8.1)].
-
14 CLINICAL STUDIES
14.1 Primary Immunization
A three-dose primary immunization series with TENIVAC was evaluated in 17 participants ages 6 to 56 years in a study conducted in Canada. [See Adverse Reactions (6.1).] The first two doses were administered two months apart, followed by a third dose six to eight months after the second dose. Serum tetanus antitoxin levels were measured by an in vivo neutralizing assay and serum diphtheria antitoxin levels were measured by an in vitro neutralizing assay. [See Clinical Pharmacology (12.1).] All 17 participants had serum tetanus and diphtheria antitoxin levels pre-vaccination and 7 days post-vaccination <0.01 IU/mL, consistent with no previous immunization. Four weeks following the second dose, all 17 participants had a serum tetanus antitoxin level >0.1 IU/mL and a serum diphtheria antitoxin level ≥0.01 IU/mL. Four weeks following the third dose, all 17 participants had a serum diphtheria antitoxin level >0.1 IU/mL.
14.2 Booster Immunization
In the US multicenter booster immunization study (TDC01) [see Adverse Reactions (6.1)], the immune response to a dose of TENIVAC was evaluated in an open-label manner in a subset of participants 11 to 59 years of age, and in comparison to DECAVAC in participants ≥60 years of age who were randomized to receive a dose of either TENIVAC or DECAVAC. Tetanus immune responses, measured by ELISA [see Clinical Pharmacology (12.1)] are presented in Table 3. Diphtheria immune responses, measured by a microneutralization assay [see Clinical Pharmacology (12.1)], are presented in Table 4.
Among adults 65 years of age and over who received TENIVAC (N = 419), 94.5% (95% confidence interval 91.9, 96.5) had a post-vaccination tetanus antitoxoid level ≥0.1 IU/mL and 61.1% (95% confidence interval 56.2, 65.8) had a post-vaccination diphtheria antitoxoid level ≥0.1 IU/mL.
Table 3: Tetanus Antitoxoid Levels and Booster Response Rates Following a Dose of TENIVAC, by Age Group, and for Adults ≥60 Years of Age, Compared to DECAVAC, per Protocol Immunogenicity Population Treatment Group Age Group Timing Percent of Participants With Specified Level of Tetanus Antitoxoid and Booster Response ≥0.1 IU/mL
% (95% CI)≥1.0 IU/mL
% (95% CI)Booster Response*
% (95% CI)Pre- indicates pre-vaccination bleed. Post- indicates 26–42 days post-vaccination bleed. - *
- Booster response: If pre-vaccination level ≤0.10 IU/mL, 4-fold increase and post-vaccination level ≥0.10 IU/mL. If pre-vaccination level >0.10 IU/mL and ≤2.7 IU/mL, 4-fold increase. If pre-vaccination level >2.7 IU/mL, 2-fold increase.
- †
- TENIVAC non-inferior to DECAVAC [upper limit of 95% CI for difference (DECAVAC minus TENIVAC) <5%].
- ‡
- Non-inferiority criteria not prospectively specified for this endpoint.
- §
- TENIVAC non-inferior to DECAVAC [upper limit of 95% CI for difference (DECAVAC minus TENIVAC) <10%].
TENIVAC Adolescents
11 to 18 years
(N = 470)Pre- 97.9
(96.1, 99.0)48.7
(44.1, 53.3)- Post- 100.0
(99.2, 100)99.8
(98.8, 100)92.8
(90.0, 94.9)Adults
19 to 59 years
(N = 237)Pre- 97.5
(94.6, 99.1)77.6
(71.8, 82.8)- Post- 100.0
(98.5, 100)99.6
(97.7, 100)84.0
(78.7, 88.4)Adults
≥60 years
(N = 661)Pre- 76.2
(72.8, 79.4)43.7
(39.9, 47.6)- Post- 96.1†
(94.3, 97.4)90.6‡
(88.1, 92.7)82.3§
(79.2, 85.1)DECAVAC Adults
≥60 years
(N = 658)Pre- 75.2
(71.7, 78.5)45.7
(41.9, 49.6)- Post- 97.3
(95.7, 98.4)91.9
(89.6, 93.9)83.7
(80.7, 86.5)Table 4: Diphtheria Antitoxin Levels and Booster Response Rates Following a Dose of TENIVAC, by Age Group, and for Adults ≥60 Years of Age, Compared to DECAVAC, per Protocol Immunogenicity Population Treatment Group Age Group Timing Percent of Participants With Specified Level of Diphtheria Antitoxin and Booster Response ≥0.01 IU/mL
% (95% CI)≥0.1 IU/mL
% (95% CI)≥1.0 IU/mL
% (95% CI)Booster Response*
% (95% CI)Pre- indicates pre-vaccination bleed. Post- indicates 26–42 days post-vaccination bleed. - *
- Booster response: If pre-vaccination level ≤0.10 IU/mL, 4-fold increase and post-vaccination level ≥0.10 IU/mL. If pre-vaccination level >0.10 IU/mL and ≤2.56 IU/mL, 4-fold increase. If pre-vaccination level >2.56 IU/mL, 2-fold increase.
- †
- Non-inferiority criteria not prospectively specified for this endpoint.
- ‡
- TENIVAC non-inferior to DECAVAC [upper limit of 95% CI for difference (DECAVAC minus TENIVAC) <10%].
TENIVAC Adolescents
11 to 18 years
(N = 470)Pre- 99.1
(97.8, 99.8)78.7
(74.7, 82.3)18.5
(15.1, 22.3)- Post- 100.0
(99.2, 100)99.8
(98.8, 100)98.9
(97.5, 99.7)95.7
(93.5, 97.4)Adults
19 to 59 years
(N = 237)Pre- 96.6
(93.5, 98.5)73.0
(66.9, 78.5)18.6
(13.8, 24.1)- Post- 99.2
(97.0, 99.9)97.5
(94.6, 99.1)91.1
(86.8, 94.4)89.9
(85.3, 93.4)Adults
≥60 years
(N = 661)Pre- 61.9
(58.1, 65.6)29.0
(25.6, 32.7)8.5
(6.5, 10.9)- Post- 88.0†
(85.3, 90.4)71.1‡
(67.5, 74.5)47.5†
(43.6, 51.4)65.5‡
(61.7, 69.1)DECAVAC Adults
≥60 years
(N = 658)Pre- 61.7
(57.9, 65.4)32.2
(28.7, 35.9)10.5
(8.3, 13.1)- Post- 87.4
(84.6, 89.8)70.7
(67.0, 74.1)45.7
(41.9, 49.6)62.9
(59.1, 66.6) -
15 REFERENCES
- 1
- CDC. Diphtheria, tetanus and pertussis: recommendations for vaccine use and other preventive measures. Recommendations of the Immunization Practices Advisory Committee (ACIP). MMWR 1991;40(RR-10):1-28.
- 2
- Stratton KR, et al, editors. Adverse events associated with childhood vaccines; evidence bearing on causality. Washington, DC: National Academy Press 1994. p. 67-117.
- 3
- Mueller JH, Miller PA. Variable factors influencing the production of tetanus toxin. J Bacteriol 1954;67(3):271-7.
- 4
- Stainer DW. Production of diphtheria toxin. In: Manclark CR, editor. Proceedings of an informal consultation on the World Health Organization requirements for diphtheria, tetanus, pertussis and combined vaccines. United States Public Health Services, Bethesda, MD. DHHS 91-1174. 1991. p. 7-11.
- 5
- FDA. Department of Health and Human Services (DHHS). Biological products; bacterial vaccines and toxoids; implementation of efficacy review; proposed rule. Fed Reg 1985;50(240):51002-117.
- 6
- Wassilak SGF, et al. Tetanus toxoid. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. 5th ed. Philadelphia, PA: W.B. Saunders Company; 2008. p. 805-39.
- 7
- Vitek CR and Wharton M. Diphtheria toxoid. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. 5th ed. Philadelphia, PA: W.B. Saunders Company; 2008. p. 139-56.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
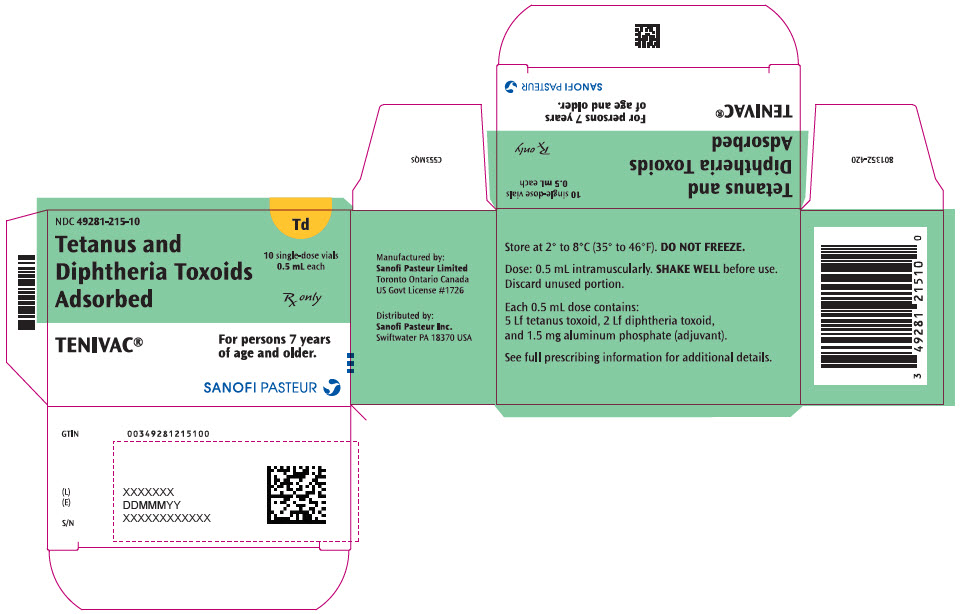
Single-dose Vial, NDC No. 49281-215-58; in package of 10 vials, NDC No. 49281-215-10. Contains no latex.
Single-dose Syringe, NDC No. 49281-215-88; in package of 10 syringes, NDC No. 49281-215-15. The tip caps of the prefilled syringes may contain natural rubber latex. No other components contain latex.
-
17 PATIENT COUNSELING INFORMATION
Before administration of TENIVAC health-care providers should inform the patient, parent or guardian of the benefits and risks of the vaccine and the importance of completing the primary immunization series or receiving recommended booster doses, as appropriate, unless a contraindication to further immunization exists.
The health-care provider should inform the patient, parent or guardian about the potential for adverse reactions that have been temporally associated with TENIVAC or other vaccines containing similar components. The health-care provider should provide the Vaccine Information Statements (VISs) which are required by the National Childhood Vaccine Injury Act of 1986 to be given with each immunization. Patients, parents, or guardians should be instructed to report adverse reactions to their health-care provider.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 0.5 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 10 Vial Package
- PRINCIPAL DISPLAY PANEL - 0.5 mL Syringe Label
- PRINCIPAL DISPLAY PANEL - 10 Syringe Package
-
INGREDIENTS AND APPEARANCE
TENIVAC
clostridium tetani toxoid antigen (formaldehyde inactivated) and corynebacterium diphtheriae toxoid antigen (formaldehyde inactivated) injection, suspensionProduct Information Product Type VACCINE Item Code (Source) NDC:49281-215 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CLOSTRIDIUM TETANI TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) (UNII: K3W1N8YP13) (CLOSTRIDIUM TETANI TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) - UNII:K3W1N8YP13) CLOSTRIDIUM TETANI TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) 5 [Lf] in 0.5 mL CORYNEBACTERIUM DIPHTHERIAE TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) (UNII: IRH51QN26H) (CORYNEBACTERIUM DIPHTHERIAE TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) - UNII:IRH51QN26H) CORYNEBACTERIUM DIPHTHERIAE TOXOID ANTIGEN (FORMALDEHYDE INACTIVATED) 2 [Lf] in 0.5 mL Inactive Ingredients Ingredient Name Strength ALUMINUM PHOSPHATE (UNII: F92V3S521O) 1.5 mg in 0.5 mL FORMALDEHYDE (UNII: 1HG84L3525) Product Characteristics Color WHITE (WHITE CLOUDY) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:49281-215-10 10 in 1 PACKAGE 1 NDC:49281-215-58 0.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:49281-215-15 10 in 1 PACKAGE 2 NDC:49281-215-88 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103171 12/08/2010 Labeler - Sanofi Pasteur Inc. (086723285) Establishment Name Address ID/FEI Business Operations Sanofi Pasteur Limited 208206623 MANUFACTURE(49281-215)