Label: STELARA- ustekinumab injection, solution
STELARA- ustekinumab solution



-
NDC Code(s):
57894-054-16,
57894-054-27,
57894-060-02,
57894-060-03, view more57894-060-04, 57894-061-02, 57894-061-03, 57894-061-04
- Packager: Janssen Biotech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated March 28, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use STELARA ®safely and effectively. See full prescribing information for STELARA ®.
STELARA ®(ustekinumab) injection, for subcutaneous or intravenous use
Initial U.S. Approval: 2009INDICATIONS AND USAGE
STELARA ®is a human interleukin-12 and -23 antagonist indicated for the treatment of:
Adult patients with:
- moderate to severe plaque psoriasis (PsO)who are candidates for phototherapy or systemic therapy. ( 1.1)
- active psoriatic arthritis (PsA). ( 1.2)
- moderately to severely active Crohn's disease (CD). ( 1.3)
- moderately to severely active ulcerative colitis.( 1.4)
Pediatric patients 6 years and older with:
DOSAGE AND ADMINISTRATION
Psoriasis Adult Subcutaneous Recommended Dosage ( 2.1) :
Weight Range (kilograms) Dose less than or equal to 100 kg 45 mg administered subcutaneously initially and 4 weeks later, followed by 45 mg administered subcutaneously every 12 weeks greater than 100 kg 90 mg administered subcutaneously initially and 4 weeks later, followed by 90 mg administered subcutaneously every 12 weeks Psoriasis Pediatric Patients (6 to 17 years old) Subcutaneous Recommended Dosage ( 2.1) :
Weight-based dosing is recommended at the initial dose, 4 weeks later, then every 12 weeks thereafter.
Weight Range (kilograms) Dose less than 60 kg 0.75 mg/kg 60 kg to 100 kg 45 mg greater than 100 kg 90 mg Psoriatic Arthritis Adult Subcutaneous Recommended Dosage ( 2.2):
- The recommended dosage is 45 mg administered subcutaneously initially and 4 weeks later, followed by 45 mg administered subcutaneously every 12 weeks.
- For patients with co-existent moderate-to-severe plaque psoriasis weighing greater than 100 kg, the recommended dosage is 90 mg administered subcutaneously initially and 4 weeks later, followed by 90 mg administered subcutaneously every 12 weeks.
Psoriatic Arthritis Pediatric (6 to 17 years old) Subcutaneous Recommended Dosage ( 2.2): Weight-based dosing is recommended at the initial dose, 4 weeks later, then every 12 weeks thereafter.
Weight Range (kilograms) Dose less than 60 kg 0.75 mg/kg 60 kg or more 45 mg greater than 100 kg with co-existent moderate-to-severe plaque psoriasis 90 mg Crohn's Disease and Ulcerative Colitis Initial Adult Intravenous Recommended Dosage ( 2.3) :
A single intravenous infusion using weight-based dosing:
Weight Range (kilograms) Recommended Dosage up to 55 kg 260 mg (2 vials) greater than 55 kg to 85 kg 390 mg (3 vials) greater than 85 kg 520 mg (4 vials) Crohn's Disease and Ulcerative Colitis Maintenance Adult Subcutaneous Recommended Dosage ( 2.3) :
A subcutaneous 90 mg dose 8 weeks after the initial intravenous dose, then every 8 weeks thereafter.
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Clinically significant hypersensitivity to ustekinumab or to any of the excipients in STELARA ®. ( 4)
WARNINGS AND PRECAUTIONS
- Infections: Serious infections have occurred. Avoid starting STELARA ®during any clinically important active infection. If a serious infection or clinically significant infection develops, discontinue STELARA ®until the infection resolves. ( 5.1)
- Theoretical Risk for Particular Infections: Serious infections from mycobacteria, salmonella and Bacillus Calmette-Guerin (BCG) vaccinations have been reported in patients genetically deficient in IL-12/IL-23. Consider diagnostic tests for these infections as dictated by clinical circumstances. ( 5.2)
- Tuberculosis (TB): Evaluate patients for TB prior to initiating treatment with STELARA ®. Initiate treatment of latent TB before administering STELARA ®. ( 5.3)
- Malignancies: STELARA ®may increase risk of malignancy. The safety of STELARA ®in patients with a history of or a known malignancy has not been evaluated. ( 5.4)
- Hypersensitivity Reactions: If an anaphylactic or other clinically significant hypersensitivity reaction occurs, institute appropriate therapy and discontinue STELARA ®( 5.5)
- Posterior Reversible Encephalopathy Syndrome (PRES): If PRES is suspected, treat promptly, and discontinue STELARA ®. ( 5.6)
- Immunizations:Avoid use of live vaccines in patients during treatment with STELARA ®. ( 5.7)
- Noninfectious Pneumonia: Cases of interstitial pneumonia, eosinophilic pneumonia, and cryptogenic organizing pneumonia have been reported during post-approval use of STELARA ®. If diagnosis is confirmed, discontinue STELARA ®and institute appropriate treatment. ( 5.8)
ADVERSE REACTIONS
Most common adverse reactions are:
- Psoriasis (≥3%):nasopharyngitis, upper respiratory tract infection, headache, and fatigue. ( 6.1)
- Crohn's Disease, induction (≥3%):vomiting. ( 6.1)
- Crohn's Disease, maintenance (≥3%):nasopharyngitis, injection site erythema, vulvovaginal candidiasis/mycotic infection, bronchitis, pruritus, urinary tract infection, and sinusitis. ( 6.1)
- Ulcerative colitis, induction (≥3%):nasopharyngitis ( 6.1)
- Ulcerative colitis, maintenance (≥3%):nasopharyngitis, headache, abdominal pain, influenza, fever, diarrhea, sinusitis, fatigue, and nausea ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Plaque Psoriasis (PsO)
1.2 Psoriatic Arthritis (PsA)
1.3 Crohn's Disease (CD)
1.4 Ulcerative Colitis
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Plaque Psoriasis
2.2 Recommended Dosage in Psoriatic Arthritis
2.3 Recommended Dosage in Crohn's Disease and Ulcerative Colitis
2.4 General Considerations for Administration
2.5 Instructions for Administration of STELARA ®Prefilled Syringes Equipped with Needle Safety Guard
2.6 Preparation and Administration of STELARA ®130 mg/26 mL (5 mg/mL) Vial for Intravenous Infusion (Crohn's Disease and Ulcerative Colitis)
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Theoretical Risk for Vulnerability to Particular Infections
5.3 Pre-treatment Evaluation for Tuberculosis
5.4 Malignancies
5.5 Hypersensitivity Reactions
5.6 Posterior Reversible Encephalopathy Syndrome (PRES)
5.7 Immunizations
5.8 Noninfectious Pneumonia
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Therapies
7.2 CYP450 Substrates
7.3 Allergen Immunotherapy
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Adult Plaque Psoriasis
14.2 Pediatric Plaque Psoriasis
14.3 Psoriatic Arthritis
14.4 Crohn's Disease
14.5 Ulcerative Colitis
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Plaque Psoriasis (PsO)
STELARA ®is indicated for the treatment of adults and pediatric patients 6 years of age and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy.
1.2 Psoriatic Arthritis (PsA)
STELARA ®is indicated for the treatment of adults and pediatric patients 6 years of age and older with active psoriatic arthritis.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Plaque Psoriasis
Subcutaneous Adult Dosage Regimen
- For patients weighing 100 kg or less, the recommended dose is 45 mg initially and 4 weeks later, followed by 45 mg every 12 weeks.
- For patients weighing more than 100 kg, the recommended dose is 90 mg initially and 4 weeks later, followed by 90 mg every 12 weeks.
In subjects weighing more than 100 kg, 45 mg was also shown to be efficacious. However, 90 mg resulted in greater efficacy in these subjects [see Clinical Studies (14)] .
Subcutaneous Pediatric Dosage Regimen
Administer STELARA ®subcutaneously at Weeks 0 and 4, then every 12 weeks thereafter.
The recommended dose of STELARA ®for pediatric patients (6–17 years old) with plaque psoriasis based on body weight is shown below (Table 1).
Table 1: Recommended Dose of STELARA ®for Subcutaneous Injection in Pediatric Patients (6–17 years old) with Plaque Psoriasis Body Weight of Patient at the Time of Dosing Recommended Dose less than 60 kg 0.75 mg/kg 60 kg to 100 kg 45 mg more than 100 kg 90 mg For pediatric patients weighing less than 60 kg, the administration volume for the recommended dose (0.75 mg/kg) is shown in Table 2; withdraw the appropriate volume from the single-dose vial.
Table 2: Injection volumes of STELARA ®45 mg/0.5 mL single-dose vials for pediatric patients (6–17 years old) with plaque psoriasis and pediatric patients (6–17 years old) with psoriatic arthritis *weighing less than 60 kg Body Weight (kg) at the time of dosing Dose (mg) Volume of injection (mL) - *
- Refer to 2.2 Psoriatic Arthritis; Subcutaneous Pediatric Dosage Regimen.
15 11.3 0.12 16 12.0 0.13 17 12.8 0.14 18 13.5 0.15 19 14.3 0.16 20 15.0 0.17 21 15.8 0.17 22 16.5 0.18 23 17.3 0.19 24 18.0 0.20 25 18.8 0.21 26 19.5 0.22 27 20.3 0.22 28 21.0 0.23 29 21.8 0.24 30 22.5 0.25 31 23.3 0.26 32 24 0.27 33 24.8 0.27 34 25.5 0.28 35 26.3 0.29 36 27 0.3 37 27.8 0.31 38 28.5 0.32 39 29.3 0.32 40 30 0.33 41 30.8 0.34 42 31.5 0.35 43 32.3 0.36 44 33 0.37 45 33.8 0.37 46 34.5 0.38 47 35.3 0.39 48 36 0.4 49 36.8 0.41 50 37.5 0.42 51 38.3 0.42 52 39 0.43 53 39.8 0.44 54 40.5 0.45 55 41.3 0.46 56 42 0.46 57 42.8 0.47 58 43.5 0.48 59 44.3 0.49 2.2 Recommended Dosage in Psoriatic Arthritis
Subcutaneous Adult Dosage Regimen
- The recommended dose is 45 mg initially and 4 weeks later, followed by 45 mg every 12 weeks.
- For patients with co-existent moderate-to-severe plaque psoriasis weighing more than 100 kg, the recommended dose is 90 mg initially and 4 weeks later, followed by 90 mg every 12 weeks.
Subcutaneous Pediatric Dosage Regimen
Administer STELARA ®subcutaneously at Weeks 0 and 4, then every 12 weeks thereafter.
The recommended dose of STELARA ®for pediatric patients (6 to 17 years old) with psoriatic arthritis, based on body weight, is shown below (Table 3).
Table 3: Recommended Dose of STELARA ®for Subcutaneous Injection in Pediatric Patients (6 to 17 years old) with Psoriatic Arthritis Body Weight of Patient at the Time of Dosing Recommended Dose - *
- For pediatric patients weighing less than 60 kg, the administration volume for the recommended dose (0.75 mg/kg) is shown in Table 2; withdraw the appropriate volume from the single-dose vial.
less than 60 kg * 0.75 mg/kg 60 kg or more 45 mg greater than 100 kg with co-existent moderate-to-severe plaque psoriasis 90 mg 2.3 Recommended Dosage in Crohn's Disease and Ulcerative Colitis
Intravenous Induction Adult Dosage Regimen
A single intravenous infusion dose of STELARA ®using the weight-based dosage regimen specified in Table 4 [see Instructions for dilution of STELARA® 130 mg vial for intravenous infusion (2.6)] .
Table 4: Initial Intravenous Dosage of STELARA ® Body Weight of Patient at the time of dosing Dose Number of 130 mg/26 mL (5 mg/mL) STELARA ®vials 55 kg or less 260 mg 2 more than 55 kg to 85 kg 390 mg 3 more than 85 kg 520 mg 4 2.4 General Considerations for Administration
- STELARA ®is intended for use under the guidance and supervision of a healthcare provider . STELARA ®should only be administered to patients who will be closely monitored and have regular follow-up visits with a healthcare provider. The appropriate dose should be determined by a healthcare provider using the patient's current weight at the time of dosing. In pediatric patients, it is recommended that STELARA ®be administered by a healthcare provider. If a healthcare provider determines that it is appropriate, a patient may self-inject or a caregiver may inject STELARA ®after proper training in subcutaneous injection technique. Instruct patients to follow the directions provided in the Medication Guide [see Medication Guide] .
- The needle cover on the prefilled syringe contains dry natural rubber (a derivative of latex). The needle cover should not be handled by persons sensitive to latex.
- It is recommended that each injection be administered at a different anatomic location (such as upper arms, gluteal regions, thighs, or any quadrant of abdomen) than the previous injection, and not into areas where the skin is tender, bruised, erythematous, or indurated. When using the single-dose vial, a 1 mL syringe with a 27 gauge, ½ inch needle is recommended.
- Prior to administration, visually inspect STELARA ®for particulate matter and discoloration. STELARA ®is a colorless to light yellow solution and may contain a few small translucent or white particles. Do not use STELARA ®if it is discolored or cloudy, or if other particulate matter is present. STELARA ®does not contain preservatives; therefore, discard any unused product remaining in the vial and/or syringe.
2.5 Instructions for Administration of STELARA ®Prefilled Syringes Equipped with Needle Safety Guard
Refer to the diagram below for the provided instructions.
To prevent premature activation of the needle safety guard, do not touch the NEEDLE GUARD ACTIVATION CLIPS at any time during use.
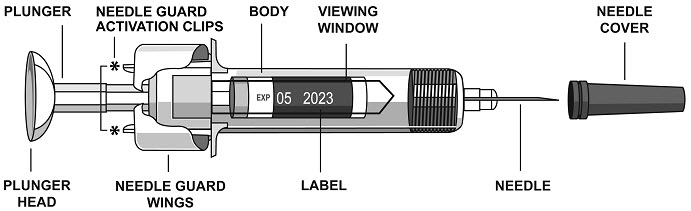
- Hold the BODY and remove the NEEDLE COVER. Do not hold the PLUNGER or PLUNGER HEAD while removing the NEEDLE COVER or the PLUNGER may move. Do not use the prefilled syringe if it is dropped without the NEEDLE COVER in place.
- Inject STELARA ®subcutaneously as recommended [see Dosage and Administration (2.1, 2.2, 2.3)] .
- Inject all of the medication by pushing in the PLUNGER until the PLUNGER HEAD is completely between the needle guard wings.
Injection of the entire prefilled syringe contents is necessary to activate the needle guard.
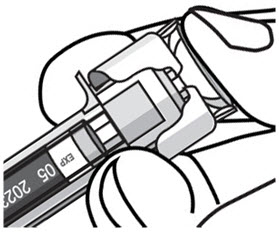
- After injection, maintain the pressure on the PLUNGER HEAD and remove the needle from the skin. Slowly take your thumb off the PLUNGER HEAD to allow the empty syringe to move up until the entire needle is covered by the needle guard, as shown by the illustration below:
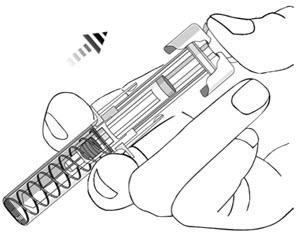
- Used syringes should be placed in a puncture-resistant container.
2.6 Preparation and Administration of STELARA ®130 mg/26 mL (5 mg/mL) Vial for Intravenous Infusion (Crohn's Disease and Ulcerative Colitis)
STELARA ®solution for intravenous infusion must be diluted, prepared and infused by a healthcare professional using aseptic technique.
- Calculate the dose and the number of STELARA ®vials needed based on patient weight (Table 4). Each 26 mL vial of STELARA ®contains 130 mg of ustekinumab.
- Withdraw, and then discard a volume of the 0.9% Sodium Chloride Injection, USP from the 250 mL infusion bag equal to the volume of STELARA ®to be added (discard 26 mL sodium chloride for each vial of STELARA ®needed, for 2 vials- discard 52 mL, for 3 vials- discard 78 mL, 4 vials- discard 104 mL). Alternatively, a 250 mL infusion bag containing 0.45% Sodium Chloride Injection, USP may be used.
- Withdraw 26 mL of STELARA ®from each vial needed and add it to the 250 mL infusion bag. The final volume in the infusion bag should be 250 mL. Gently mix.
- Visually inspect the diluted solution before infusion. Do not use if visibly opaque particles, discoloration or foreign particles are observed.
- Infuse the diluted solution over a period of at least one hour. Once diluted, the infusion should be completely administered within eight hours of the dilution in the infusion bag.
- Use only an infusion set with an in-line, sterile, non-pyrogenic, low protein-binding filter (pore size 0.2 micrometer).
- Do not infuse STELARA ®concomitantly in the same intravenous line with other agents.
- STELARA ®does not contain preservatives. Each vial is for a single-dose only. Discard any remaining solution. Dispose any unused medicinal product in accordance with local requirements.
Storage
If necessary, the diluted infusion solution may be kept at room temperature up to 25°C (77°F) for up to 7 hours. Storage time at room temperature begins once the diluted solution has been prepared. The infusion should be completed within 8 hours after the dilution in the infusion bag (cumulative time after preparation including the storage and the infusion period). Do not freeze. Discard any unused portion of the infusion solution.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
STELARA ®is contraindicated in patients with clinically significant hypersensitivity to ustekinumab or to any of the excipients in STELARA ®[see Warnings and Precautions (5.5)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
STELARA ®may increase the risk of infections and reactivation of latent infections. Serious bacterial, mycobacterial, fungal, and viral infections were observed in patients receiving STELARA ®[see Adverse Reactions (6.1, 6.3)] .
Serious infections requiring hospitalization, or otherwise clinically significant infections, reported in clinical trials included the following:
- Plaque Psoriasis: diverticulitis, cellulitis, pneumonia, appendicitis, cholecystitis, sepsis, osteomyelitis, viral infections, gastroenteritis, and urinary tract infections.
- Psoriatic arthritis: cholecystitis.
- Crohn's disease: anal abscess, gastroenteritis, ophthalmic herpes zoster, pneumonia, and listeria meningitis.
- Ulcerative colitis: gastroenteritis, ophthalmic herpes zoster, pneumonia, and listeriosis.
Avoid initiating treatment with STELARA ®in patients with any clinically important active infection until the infection resolves or is adequately treated. Consider the risks and benefits of treatment prior to initiating use of STELARA ®in patients with a chronic infection or a history of recurrent infection.
Instruct patients to seek medical advice if signs or symptoms suggestive of an infection occur while on treatment with STELARA ®and discontinue STELARA ®for serious or clinically significant infections until the infection resolves or is adequately treated.
5.2 Theoretical Risk for Vulnerability to Particular Infections
Individuals genetically deficient in IL-12/IL-23 are particularly vulnerable to disseminated infections from mycobacteria (including nontuberculous, environmental mycobacteria), salmonella (including nontyphi strains), and Bacillus Calmette-Guerin (BCG) vaccinations. Serious infections and fatal outcomes have been reported in such patients.
It is not known whether patients with pharmacologic blockade of IL-12/IL-23 from treatment with STELARA ®may be susceptible to these types of infections. Consider appropriate diagnostic testing, (e.g., tissue culture, stool culture, as dictated by clinical circumstances).
5.3 Pre-treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis infection prior to initiating treatment with STELARA ®.
Avoid administering STELARA ®to patients with active tuberculosis infection. Initiate treatment of latent tuberculosis prior to administering STELARA ®. Consider anti-tuberculosis therapy prior to initiation of STELARA ®in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed. Closely monitor patients receiving STELARA ®for signs and symptoms of active tuberculosis during and after treatment.
5.4 Malignancies
STELARA ®is an immunosuppressant and may increase the risk of malignancy. Malignancies were reported among subjects who received STELARA ®in clinical trials [see Adverse Reactions (6.1)] . In rodent models, inhibition of IL-12/IL-23p40 increased the risk of malignancy [see Nonclinical Toxicology (13)] .
The safety of STELARA ®has not been evaluated in patients who have a history of malignancy or who have a known malignancy.
There have been post-marketing reports of the rapid appearance of multiple cutaneous squamous cell carcinomas in patients receiving STELARA ®who had pre-existing risk factors for developing non-melanoma skin cancer. Monitor all patients receiving STELARA ®for the appearance of non-melanoma skin cancer. Closely follow patients greater than 60 years of age, those with a medical history of prolonged immunosuppressant therapy and those with a history of PUVA treatment [see Adverse Reactions (6.1)] .
5.5 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis and angioedema, have been reported with STELARA ®[see Adverse Reactions (6.1, 6.3)] . If an anaphylactic or other clinically significant hypersensitivity reaction occurs, institute appropriate therapy and discontinue STELARA ®.
5.6 Posterior Reversible Encephalopathy Syndrome (PRES)
Two cases of posterior reversible encephalopathy syndrome (PRES), also known as Reversible Posterior Leukoencephalopathy Syndrome (RPLS), were reported in clinical trials. Cases have also been reported in postmarketing experience in patients with psoriasis, psoriatic arthritis and Crohn's disease. Clinical presentation included headaches, seizures, confusion, visual disturbances, and imaging changes consistent with PRES a few days to several months after ustekinumab initiation. A few cases reported latency of a year or longer. Patients recovered with supportive care following withdrawal of ustekinumab.
Monitor all patients treated with STELARA ®for signs and symptoms of PRES. If PRES is suspected, promptly administer appropriate treatment and discontinue STELARA ®.
5.7 Immunizations
Prior to initiating therapy with STELARA ®, patients should receive all age-appropriate immunizations as recommended by current immunization guidelines. Patients being treated with STELARA ®should avoid receiving live vaccines. Avoid administering BCG vaccines during treatment with STELARA ®or for one year prior to initiating treatment or one year following discontinuation of treatment. Caution is advised when administering live vaccines to household contacts of patients receiving STELARA ®because of the potential risk for shedding from the household contact and transmission to patient.
Non-live vaccinations received during a course of STELARA ®may not elicit an immune response sufficient to prevent disease.
5.8 Noninfectious Pneumonia
Cases of interstitial pneumonia, eosinophilic pneumonia and cryptogenic organizing pneumonia have been reported during post-approval use of STELARA ®. Clinical presentations included cough, dyspnea, and interstitial infiltrates following one to three doses. Serious outcomes have included respiratory failure and prolonged hospitalization. Patients improved with discontinuation of therapy and in certain cases administration of corticosteroids. If diagnosis is confirmed, discontinue STELARA ®and institute appropriate treatment [see Postmarketing Experience (6.3)] .
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the label:
- Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.5)]
- Posterior Reversible Encephalopathy Syndrome (PRES) [see Warnings and Precautions (5.6)]
- Noninfectious Pneumonia [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Subjects with Plaque Psoriasis
The safety data reflect exposure to STELARA ®in 3117 adult subjects with plaque psoriasis, including 2414 exposed for at least 6 months, 1855 exposed for at least one year, 1653 exposed for at least two years, 1569 exposed for at least three years, 1482 exposed for at least four years and 838 exposed for at least five years.
Table 5 summarizes the adverse reactions that occurred at a rate of at least 1% with higher rates in the STELARA ®groups during the placebo-controlled period of Ps STUDY 1 and Ps STUDY 2 [see Clinical Studies (14)] .
Table 5: Adverse Reactions, Reported by ≥1% of Subjects with Plaque Psoriasis and at Higher Rates in the STELARA ®groups through Week 12 in Ps STUDY 1 and Ps STUDY 2 STELARA ® Placebo 45 mg 90 mg Subjects treated 665 664 666 Nasopharyngitis 51 (8%) 56 (8%) 49 (7%) Upper respiratory tract infection 30 (5%) 36 (5%) 28 (4%) Headache 23 (3%) 33 (5%) 32 (5%) Fatigue 14 (2%) 18 (3%) 17 (3%) Back pain 8 (1%) 9 (1%) 14 (2%) Dizziness 8 (1%) 8 (1%) 14 (2%) Pharyngolaryngeal pain 7 (1%) 9 (1%) 12 (2%) Pruritus 9 (1%) 10 (2%) 9 (1%) Injection site erythema 3 (<1%) 6 (1%) 13 (2%) Myalgia 4 (1%) 7 (1%) 8 (1%) Depression 3 (<1%) 8 (1%) 4 (1%) Adverse reactions that occurred at rates less than 1% in the controlled period of Ps STUDIES 1 and 2 through week 12 included: cellulitis, herpes zoster, diverticulitis, and certain injection site reactions (pain, swelling, pruritus, induration, hemorrhage, bruising, and irritation).
One case of PRES occurred during adult plaque psoriasis clinical trials [see Warnings and Precautions (5.6)] .
Infections
In the placebo-controlled period of clinical trials of subjects with plaque psoriasis (average follow-up of 12.6 weeks for placebo-treated subjects and 13.4 weeks for STELARA ®-treated subjects), 27% of STELARA ®-treated subjects reported infections (1.39 per subject-year of follow-up) compared with 24% of placebo-treated subjects (1.21 per subject-year of follow-up). Serious infections occurred in 0.3% of STELARA ®-treated subjects (0.01 per subject-year of follow-up) and in 0.4% of placebo-treated subjects (0.02 per subject-year of follow-up) [see Warnings and Precautions (5.1)] .
In the controlled and non-controlled portions of plaque psoriasis clinical trials (median follow-up of 3.2 years), representing 8998 subject-years of exposure, 72.3% of STELARA ®-treated subjects reported infections (0.87 per subject-years of follow-up). Serious infections were reported in 2.8% of subjects (0.01 per subject-years of follow-up).
Malignancies
In the controlled and non-controlled portions of plaque psoriasis clinical trials (median follow-up of 3.2 years, representing 8998 subject-years of exposure), 1.7% of STELARA ®-treated subjects reported malignancies excluding non-melanoma skin cancers (0.60 per hundred subject-years of follow-up). Non-melanoma skin cancer was reported in 1.5% of STELARA ®-treated subjects (0.52 per hundred subject-years of follow-up) [see Warnings and Precautions (5.4)] . The most frequently observed malignancies other than non-melanoma skin cancer during the clinical trials were: prostate, melanoma, colorectal and breast. Malignancies other than non-melanoma skin cancer in STELARA ®-treated patients during the controlled and uncontrolled portions of trials were similar in type and number to what would be expected in the general U.S. population according to the SEER database (adjusted for age, gender and race). 1
Pediatric Subjects with Plaque Psoriasis
The safety of STELARA ®was assessed in two trials of pediatric subjects with moderate to severe plaque psoriasis. Ps STUDY 3 evaluated safety for up to 60 weeks in 110 pediatric subjects 12 to 17 years old. Ps STUDY 4 evaluated safety for up to 56 weeks in 44 pediatric subjects 6 to 11 years old. The safety profile in pediatric subjects was similar to the safety profile from trials in adults with plaque psoriasis.
Psoriatic Arthritis
The safety of STELARA ®was assessed in 927 subjects in two randomized, double-blind, placebo-controlled trials in adults with active psoriatic arthritis (PsA). The overall safety profile of STELARA ®in subjects with PsA was consistent with the safety profile seen in adult psoriasis clinical trials. A higher incidence of arthralgia, nausea, and dental infections was observed in STELARA ®-treated subjects when compared with placebo-treated subjects (3% vs. 1% for arthralgia and 3% vs. 1% for nausea; 1% vs. 0.6% for dental infections) in the placebo-controlled portions of the PsA clinical trials.
Crohn's Disease
The safety of STELARA ®was assessed in 1407 subjects with moderately to severely active Crohn's disease (Crohn's Disease Activity Index [CDAI] greater than or equal to 220 and less than or equal to 450) in three randomized, double-blind, placebo-controlled, parallel-group, multicenter trials. These 1407 subjects included 40 subjects who received a prior investigational intravenous ustekinumab formulation but were not included in the efficacy analyses. In trials CD-1 and CD-2 there were 470 subjects who received STELARA ®6 mg/kg as a weight-based single intravenous induction dose and 466 who received placebo [see Dosage and Administration (2.3)] . Subjects who were responders in either trial CD-1 or CD-2 were randomized to receive a subcutaneous maintenance regimen of either 90 mg STELARA ®every 8 weeks, or placebo for 44 weeks in trial CD-3. Subjects in these 3 trials may have received other concomitant therapies including aminosalicylates, immunomodulatory agents [azathioprine (AZA), 6-mercaptopurine (6-MP), methotrexate (MTX)], oral corticosteroids (prednisone or budesonide), and/or antibiotics for their Crohn's disease [see Clinical Studies (14.4)] .
The overall safety profile of STELARA ®was consistent with the safety profile seen in the adult psoriasis and psoriatic arthritis clinical trials. Common adverse reactions in trials CD-1 and CD-2 and in trial CD-3 are listed in Tables 6 and 7, respectively.
Table 6: Common adverse reactions through Week 8 in Trials CD-1 and CD-2 occurring in ≥3% of STELARA ®-treated subjects and higher than placebo Placebo STELARA ®
6 mg/kg single intravenous induction doseN=466 N=470 Vomiting 3% 4% Other less common adverse reactions reported in subjects in trials CD-1 and CD-2 included asthenia (1% vs 0.4%), acne (1% vs 0.4%), and pruritus (2% vs 0.4%).
Table 7: Common adverse reactions through Week 44 in Trial CD-3 occurring in ≥3% of STELARA ®-treated subjects and higher than placebo Placebo STELARA ®
90 mg subcutaneous maintenance dose every 8 weeksN=133 N=131 Nasopharyngitis 8% 11% Injection site erythema 0 5% Vulvovaginal candidiasis/mycotic infection 1% 5% Bronchitis 3% 5% Pruritus 2% 4% Urinary tract infection 2% 4% Sinusitis 2% 3% Infections
In patients with Crohn's disease, serious or other clinically significant infections included anal abscess, gastroenteritis, and pneumonia. In addition, listeria meningitis and ophthalmic herpes zoster were reported in one patient each [see Warnings and Precautions (5.1)] .
Malignancies
With up to one year of treatment in the Crohn's disease clinical trials, 0.2% of STELARA ®-treated subjects (0.36 events per hundred patient-years) and 0.2% of placebo-treated subjects (0.58 events per hundred patient-years) developed non-melanoma skin cancer. Malignancies other than non-melanoma skin cancers occurred in 0.2% of STELARA ®-treated subjects (0.27 events per hundred patient-years) and in none of the placebo-treated subjects.
Hypersensitivity Reactions Including Anaphylaxis
In CD trials, two patients reported hypersensitivity reactions following STELARA ®administration. One patient experienced signs and symptoms consistent with anaphylaxis (tightness of the throat, shortness of breath, and flushing) after a single subcutaneous administration (0.1% of patients receiving subcutaneous STELARA ®). In addition, one patient experienced signs and symptoms consistent with or related to a hypersensitivity reaction (chest discomfort, flushing, urticaria, and increased body temperature) after the initial intravenous STELARA ®dose (0.08% of patients receiving intravenous STELARA ®). These patients were treated with oral antihistamines or corticosteroids and in both cases symptoms resolved within an hour.
Ulcerative Colitis
The safety of STELARA ®was evaluated in two randomized, double-blind, placebo-controlled clinical trials (UC-1 [IV induction] and UC-2 [SC maintenance]) in 960 adult subjects with moderately to severely active ulcerative colitis [see Clinical Studies (14.5)] . The overall safety profile of STELARA ®in patients with ulcerative colitis was consistent with the safety profile seen across all approved indications. Adverse reactions reported in at least 3% of STELARA ®-treated subjects and at a higher rate than placebo were:
- Induction (UC-1): nasopharyngitis (7% vs 4%).
- Maintenance (UC-2): nasopharyngitis (24% vs 20%), headache (10% vs 4%), abdominal pain (7% vs 3%), influenza (6% vs 5%), fever (5% vs. 4%), diarrhea (4% vs 1%), sinusitis (4% vs 1%), fatigue (4% vs 2%), and nausea (3% vs 2%).
Infections
In patients with ulcerative colitis, serious or other clinically significant infections included gastroenteritis and pneumonia. In addition, listeriosis and ophthalmic herpes zoster were reported in one patient each [see Warnings and Precautions (5.1)] .
Malignancies
With up to one year of treatment in the ulcerative colitis clinical trials, 0.4% of STELARA ®-treated subjects (0.48 events per hundred patient-years) and 0.0% of placebo-treated subjects (0.00 events per hundred patient-years) developed non-melanoma skin cancer. Malignancies other than non-melanoma skin cancers occurred in 0.5% of STELARA ®-treated subjects (0.64 events per hundred patient-years) and 0.2% of placebo-treated subjects (0.40 events per hundred patient-years).
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of the incidence of antibodies to ustekinumab in the trials described below with the incidence of antibodies to other products may be misleading.
Approximately 6 to 12.4% of subjects treated with STELARA ®in plaque psoriasis and psoriatic arthritis clinical trials developed antibodies to ustekinumab, which were generally low-titer. In plaque psoriasis clinical trials, antibodies to ustekinumab were associated with reduced or undetectable serum ustekinumab concentrations and reduced efficacy. In plaque psoriasis trials, the majority of subjects who were positive for antibodies to ustekinumab had neutralizing antibodies.
In Crohn's disease and ulcerative colitis clinical trials, 2.9% and 4.6% of subjects, respectively, developed antibodies to ustekinumab when treated with STELARA ®for approximately one year. No apparent association between the development of antibodies to ustekinumab and the development of injection site reactions was seen.
6.3 Postmarketing Experience
The following adverse reactions have been reported during post-approval use of STELARA ®. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to STELARA ®exposure.
Immune system disorders:Serious hypersensitivity reactions (including anaphylaxis and angioedema), other hypersensitivity reactions (including rash and urticaria) .
Infections and infestations:Lower respiratory tract infection (including opportunistic fungal infections and tuberculosis).
Neurological disorders:Posterior Reversible Encephalopathy Syndrome (PRES) .
Respiratory, thoracic and mediastinal disorders:Interstitial pneumonia, eosinophilic pneumonia, and cryptogenic organizing pneumonia.
Skin reactions: Pustular psoriasis, erythrodermic psoriasis, hypersensitivity vasculitis.
-
7 DRUG INTERACTIONS
7.1 Concomitant Therapies
In plaque psoriasis trials the safety of STELARA ®in combination with immunosuppressive agents or phototherapy has not been evaluated . In psoriatic arthritis trials, concomitant MTX use did not appear to influence the safety or efficacy of STELARA ®. In Crohn's disease and ulcerative colitis induction trials, immunomodulators (6-MP, AZA, MTX) were used concomitantly in approximately 30% of subjects and corticosteroids were used concomitantly in approximately 40% and 50% of Crohn's disease and ulcerative colitis subjects, respectively. Use of these concomitant therapies did not appear to influence the overall safety or efficacy of STELARA ®.
7.2 CYP450 Substrates
The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g., IL-1, IL-6, IL-10, TNFα, IFN) during chronic inflammation. Thus, STELARA ®, an antagonist of IL-12 and IL-23, could normalize the formation of CYP450 enzymes. Upon initiation of STELARA ®in patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index, consider monitoring for therapeutic effect (e.g., for warfarin) or drug concentration (e.g., for cyclosporine) and adjust the individual dose of the drug as needed [see Clinical Pharmacology (12.3)] .
7.3 Allergen Immunotherapy
STELARA ®has not been evaluated in patients who have undergone allergy immunotherapy. STELARA ®may decrease the protective effect of allergen immunotherapy (decrease tolerance) which may increase the risk of an allergic reaction to a dose of allergen immunotherapy. Therefore, caution should be exercised in patients receiving or who have received allergen immunotherapy, particularly for anaphylaxis.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data from observational studies, published case reports, and postmarketing surveillance on the use of STELARA ®during pregnancy are insufficient to inform a drug associated risk of major birth defects, miscarriage, and other adverse maternal or fetal outcomes . Transport of human IgG antibody across the placenta increases as pregnancy progresses and peaks during the third trimester; therefore, STELARA ®may be transferred to the developing fetus (see Clinical Considerations). In animal reproductive and developmental toxicity studies, no adverse developmental effects were observed in offspring after administration of ustekinumab to pregnant monkeys at exposures greater than 100 times the maximum recommended human dose (MRHD).
The background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage of clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Because ustekinumab may theoretically interfere with immune response to infections, consider risks and benefits prior to administering live vaccines to infants exposed to STELARA ®in utero. There are insufficient data regarding exposed infant serum levels of ustekinumab at birth and the duration of persistence of ustekinumab in infant serum after birth. Although a specific timeframe to delay administration of live attenuated vaccines in infants exposed in uterois unknown, consider the risks and benefits of delaying a minimum of 6 months after birth because of the clearance of the product.
Data
Animal Data
Ustekinumab was tested in two embryo-fetal development toxicity studies in cynomolgus monkeys. No teratogenic or other adverse developmental effects were observed in fetuses from pregnant monkeys that were administered ustekinumab subcutaneously twice weekly or intravenously weekly during the period of organogenesis. Serum concentrations of ustekinumab in pregnant monkeys were greater than 100 times the serum concentration in patients treated subcutaneously with 90 mg of ustekinumab weekly for 4 weeks.
In a combined embryo-fetal development and pre- and post-natal development toxicity study, pregnant cynomolgus monkeys were administered subcutaneous doses of ustekinumab twice weekly at exposures greater than 100 times the MRHD from the beginning of organogenesis to Day 33 after delivery. Neonatal deaths occurred in the offspring of one monkey administered ustekinumab at 22.5 mg/kg and one monkey dosed at 45 mg/kg. No ustekinumab-related effects on functional, morphological, or immunological development were observed in the neonates from birth through six months of age.
8.2 Lactation
Risk Summary
Limited data from published literature suggests that ustekinumab is present in human breast milk. There are no available data on the effects of ustekinumab on milk production. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to ustekinumab are unknown. No adverse effects on the breastfed infant causally related to ustekinumab have been identified in the published literature or postmarketing experience.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for STELARA ®and any potential adverse effects on the breastfed child from STELARA ®or from the underlying maternal condition.
8.4 Pediatric Use
Plaque Psoriasis
The safety and effectiveness of STELARA ®have been established for the treatment of moderate to severe plaque psoriasis in pediatric patients 6 to 17 years of age who are candidates for phototherapy or systemic therapy.
Use of STELARA ®in pediatric patients 12 to less than 17 years of age is supported by evidence from a multicenter, randomized, 60week trial (Ps STUDY 3) that included a 12week, double-blind, placebo-controlled, parallel group portion, in 110 pediatric subjects 12 years of age and older [see Adverse Reactions (6.1), Clinical Studies (14.2)] .
Use of STELARA ®in pediatric patients 6 to 11 years of age is supported by evidence from an open-label, single-arm, efficacy, safety, and pharmacokinetics trial (Ps STUDY 4) in 44 subjects [see Adverse Reactions (6.1), Pharmacokinetics (12.3)] .
The safety and effectiveness of STELARA ®have not been established in pediatric patients less than 6 years of age with plaque psoriasis.
Psoriatic Arthritis
The safety and effectiveness of STELARA ®have been established for treatment of psoriatic arthritis in pediatric patients 6 to 17 years old.
Use of STELARA ®in these age groups is supported by evidence from adequate and well controlled trials of STELARA ®in adults with psoriasis and PsA, pharmacokinetic data from adult patients with psoriasis, adult patients with PsA and pediatric patients with psoriasis, and safety data from two clinical trials in 44 pediatric patients 6 to 11 years old with psoriasis and 110 pediatric patients 12 to 17 years old with psoriasis. The observed pre-dose (trough) concentrations are generally comparable between adult patients with psoriasis, adult patients with PsA and pediatric patients with psoriasis, and the PK exposure is expected to be comparable between adult and pediatric patients with PsA [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1, 14.2, 14.3)].
The safety and effectiveness of STELARA ®have not been established in pediatric patients less than 6 years old with psoriatic arthritis.
8.5 Geriatric Use
Of the 6709 patients exposed to STELARA ®, a total of 340 were 65 years of age or older (183 patients with plaque psoriasis, 65 patients with psoriatic arthritis, 58 patients with Crohn's disease and 34 patients with ulcerative colitis), and 40 patients were 75 years of age or older., Clinical trials of STELARA ®did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
-
10 OVERDOSAGE
Single doses up to 6 mg/kg intravenously have been administered in clinical trials without dose-limiting toxicity. In case of overdosage, monitor the patient for any signs or symptoms of adverse reactions or effects and institute appropriate symptomatic treatment immediately. Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
-
11 DESCRIPTION
Ustekinumab, a human IgG1κ monoclonal antibody, is a human interleukin-12 and -23 antagonist. Using DNA recombinant technology, ustekinumab is produced in a murine cell line (Sp2/0). The manufacturing process contains steps for the clearance of viruses. Ustekinumab is comprised of 1326 amino acids and has an estimated molecular mass that ranges from 148,079 to 149,690 Daltons.
STELARA ®(ustekinumab) injection is a sterile, preservative-free, colorless to light yellow solution and may contain a few small translucent or white particles with pH of 5.7– 6.3.
STELARA ®for Subcutaneous Use
Available as 45 mg of ustekinumab in 0.5 mL and 90 mg of ustekinumab in 1 mL, supplied as a sterile solution in a single-dose prefilled syringe with a 27 gauge fixed ½ inch needle and as 45 mg of ustekinumab in 0.5 mL in a single-dose 2 mL Type I glass vial with a coated stopper. The syringe is fitted with a passive needle guard and a needle cover that contains dry natural rubber (a derivative of latex).
Each 0.5 mL prefilled syringe or vial delivers 45 mg ustekinumab, L-histidine and L-histidine monohydrochloride monohydrate (0.5 mg), Polysorbate 80 (0.02 mg), and sucrose (38 mg).
Each 1 mL prefilled syringe delivers 90 mg ustekinumab, L-histidine and L-histidine monohydrochloride monohydrate (1 mg), Polysorbate 80 (0.04 mg), and sucrose (76 mg).
STELARA ®for Intravenous Infusion
Available as 130 mg of ustekinumab in 26 mL, supplied as a single-dose 30 mL Type I glass vial with a coated stopper.
Each 26 mL vial delivers 130 mg ustekinumab, EDTA disodium salt dihydrate (0.52 mg), L-histidine (20 mg), L-histidine hydrochloride monohydrate (27 mg), L-methionine (10.4 mg), Polysorbate 80 (10.4 mg), and sucrose (2210 mg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ustekinumab is a human IgG1қ monoclonal antibody that binds with specificity to the p40 protein subunit used by both the IL-12 and IL-23 cytokines. IL-12 and IL-23 are naturally occurring cytokines that are involved in inflammatory and immune responses, such as natural killer cell activation and CD4+ T-cell differentiation and activation. In in vitromodels, ustekinumab was shown to disrupt IL-12 and IL-23 mediated signaling and cytokine cascades by disrupting the interaction of these cytokines with a shared cell-surface receptor chain, IL-12Rβ1. The cytokines IL-12 and IL-23 have been implicated as important contributors to the chronic inflammation that is a hallmark of Crohn's disease and ulcerative colitis. In animal models of colitis, genetic absence or antibody blockade of the p40 subunit of IL-12 and IL-23, the target of ustekinumab, was shown to be protective.
12.2 Pharmacodynamics
Plaque Psoriasis
In a small exploratory trial, a decrease was observed in the expression of mRNA of its molecular targets IL-12 and IL-23 in lesional skin biopsies measured at baseline and up to two weeks post-treatment in subjects with plaque psoriasis.
Ulcerative Colitis
In both trial UC-1 (induction) and trial UC-2 (maintenance), a positive relationship was observed between exposure and rates of clinical remission, clinical response, and endoscopic improvement. The response rate approached a plateau at the ustekinumab exposures associated with the recommended dosing regimen for maintenance treatment [see Clinical Studies (14.5)] .
12.3 Pharmacokinetics
Absorption
In adult subjects with plaque psoriasis, the median time to reach the maximum serum concentration (T max) was 13.5 days and 7 days, respectively, after a single subcutaneous administration of 45 mg (N=22) and 90 mg (N=24) of ustekinumab. In healthy subjects (N=30), the median T maxvalue (8.5 days) following a single subcutaneous administration of 90 mg of ustekinumab was comparable to that observed in subjects with plaque psoriasis.
Following multiple subcutaneous doses of STELARA ®in adult subjects with plaque psoriasis, steady-state serum concentrations of ustekinumab were achieved by Week 28. The mean (±SD) steady-state trough serum ustekinumab concentrations were 0.69 ± 0.69 mcg/mL for patients less than or equal to 100 kg receiving a 45 mg dose and 0.74 ± 0.78 mcg/mL for patients greater than 100 kg receiving a 90 mg dose. There was no apparent accumulation in serum ustekinumab concentration over time when given subcutaneously every 12 weeks.
Following the recommended intravenous induction dose, mean ±SD peak serum ustekinumab concentration was 125.2 ± 33.6 mcg/mL in patients with Crohn's disease, and 129.1 ± 27.6 mcg/mL in patients with ulcerative colitis. Starting at Week 8, the recommended subcutaneous maintenance dosing of 90 mg ustekinumab was administered every 8 weeks. Steady state ustekinumab concentration was achieved by the start of the second maintenance dose. There was no apparent accumulation in ustekinumab concentration over time when given subcutaneously every 8 weeks. Mean ±SD steady-state trough concentration was 2.5 ± 2.1 mcg/mL in patients with Crohn's disease, and 3.3 ± 2.3 mcg/mL in patients with ulcerative colitis for 90 mg ustekinumab administered every 8 weeks.
Distribution
Population pharmacokinetic analyses showed that the volume of distribution of ustekinumab in the central compartment was 2.7 L (95% CI: 2.69, 2.78) in patients with Crohn's disease and 3.0 L (95% CI: 2.96, 3.07) in patients with ulcerative colitis. The total volume of distribution at steady-state was 4.6 L in patients with Crohn's disease and 4.4 L in patients with ulcerative colitis.
Elimination
The mean (±SD) half-life ranged from 14.9 ± 4.6 to 45.6 ± 80.2 days across all plaque psoriasis trials following subcutaneous administration. Population pharmacokinetic analyses showed that the clearance of ustekinumab was 0.19 L/day (95% CI: 0.185, 0.197) in patients with Crohn's disease and 0.19 L/day (95% CI: 0.179, 0.192) in patients with ulcerative colitis with an estimated median terminal half-life of approximately 19 days for both IBD (Crohn's disease and ulcerative colitis) populations.
These results indicate the pharmacokinetics of ustekinumab were similar between patients with Crohn's disease and ulcerative colitis.
Metabolism
The metabolic pathway of ustekinumab has not been characterized. As a human IgG1κ monoclonal antibody, ustekinumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Specific Populations
Weight
When given the same dose, subjects with plaque psoriasis or psoriatic arthritis weighing more than 100 kg had lower median serum ustekinumab concentrations compared with those subjects weighing 100 kg or less. The median trough serum concentrations of ustekinumab in subjects of higher weight (greater than 100 kg) in the 90 mg group were comparable to those in subjects of lower weight (100 kg or less) in the 45 mg group.
Age: Geriatric Population
A population pharmacokinetic analysis (N=106/1937 patients with plaque psoriasis greater than or equal to 65 years old) was performed to evaluate the effect of age on the pharmacokinetics of ustekinumab. There were no apparent changes in pharmacokinetic parameters (clearance and volume of distribution) in subjects older than 65 years old.
Age: Pediatric Population
Following multiple recommended doses of STELARA ®in pediatric subjects 6 to 17 years of age with plaque psoriasis, steady-state serum concentrations of ustekinumab were achieved by Week 28. At Week 28, the mean ±SD steady-state trough serum ustekinumab concentrations were 0.36 ± 0.26 mcg/mL and 0.54 ± 0.43 mcg/mL, respectively, in pediatric subjects 6 to 11 years of age and pediatric subjects 12 to 17 years of age.
Overall, the observed steady-state ustekinumab trough concentrations in pediatric patients with plaque psoriasis were within the range of those observed for adult patients with plaque psoriasis and adult patients with PsA after administration of STELARA ®.
Drug Interaction Studies
The effects of IL-12 or IL-23 on the regulation of CYP450 enzymes were evaluated in an in vitrostudy using human hepatocytes, which showed that IL-12 and/or IL-23 at levels of 10 ng/mL did not alter human CYP450 enzyme activities (CYP1A2, 2B6, 2C9, 2C19, 2D6, or 3A4). However, the clinical relevance of in vitrodata has not been established [see Drug Interactions (7.3)] .
No in vivodrug interaction studies have been conducted with STELARA ®.
Population pharmacokinetic analyses indicated that the clearance of ustekinumab was not impacted by concomitant MTX, NSAIDs, and oral corticosteroids, or prior exposure to a TNF blocker in patients with psoriatic arthritis.
In patients with Crohn's disease and ulcerative colitis, population pharmacokinetic analyses did not indicate changes in ustekinumab clearance with concomitant use of corticosteroids or immunomodulators (AZA, 6-MP, or MTX); and serum ustekinumab concentrations were not impacted by concomitant use of these medications.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential of STELARA ®. Published literature showed that administration of murine IL-12 caused an anti-tumor effect in mice that contained transplanted tumors and IL-12/IL-23p40 knockout mice or mice treated with anti-IL-12/IL-23p40 antibody had decreased host defense to tumors. Mice genetically manipulated to be deficient in both IL-12 and IL-23 or IL-12 alone developed UV-induced skin cancers earlier and more frequently compared to wild-type mice. The relevance of these experimental findings in mouse models for malignancy risk in humans is unknown.
No effects on fertility were observed in male cynomolgus monkeys that were administered ustekinumab at subcutaneous doses up to 45 mg/kg twice weekly (45 times the MRHD on a mg/kg basis) prior to and during the mating period. However, fertility and pregnancy outcomes were not evaluated in mated females.
No effects on fertility were observed in female mice that were administered an analogous IL-12/IL-23p40 antibody by subcutaneous administration at doses up to 50 mg/kg, twice weekly, prior to and during early pregnancy.
-
14 CLINICAL STUDIES
14.1 Adult Plaque Psoriasis
Two multicenter, randomized, double-blind, placebo-controlled trials (Ps STUDY 1 and Ps STUDY 2) enrolled a total of 1996 subjects 18 years of age and older with plaque psoriasis who had a minimum body surface area involvement of 10%, and Psoriasis Area and Severity Index (PASI) score ≥12, and who were candidates for phototherapy or systemic therapy. Subjects with guttate, erythrodermic, or pustular psoriasis were excluded from the trials.
Ps STUDY 1 enrolled 766 subjects and Ps STUDY 2 enrolled 1230 subjects. The trials had the same design through Week 28. In both trials, subjects were randomized in equal proportion to placebo, 45 mg or 90 mg of STELARA ®. Subjects randomized to STELARA ®received 45 mg or 90 mg doses, regardless of weight, at Weeks 0, 4, and 16. Subjects randomized to receive placebo at Weeks 0 and 4 crossed over to receive STELARA ®(either 45 mg or 90 mg) at Weeks 12 and 16.
In both trials, subjects in all treatment groups had a median baseline PASI score ranging from approximately 17 to 18. Baseline PGA score was marked or severe in 44% of subjects in Ps STUDY 1 and 40% of subjects in Ps STUDY 2. Approximately two-thirds of all subjects had received prior phototherapy, 69% had received either prior conventional systemic or biologic therapy for the treatment of psoriasis, with 56% receiving prior conventional systemic therapy and 43% receiving prior biologic therapy. A total of 28% of subjects had a history of psoriatic arthritis.
In bothtrials, the endpoints were the proportion of subjects who achieved at least a 75% reduction in PASI score (PASI 75) from baseline to Week 12 and treatment success (cleared or minimal) on the Physician's Global Assessment (PGA). The PGA is a 6-category scale ranging from 0 (cleared) to 5 (severe) that indicates the physician's overall assessment of psoriasis focusing on plaque thickness/induration, erythema, and scaling.
Clinical Response
The results of Ps STUDY 1 and Ps STUDY 2 are presented in Table 8 below.
Table 8: Clinical Outcomes at Week 12 in Adults with Plaque Psoriasis in Ps STUDY 1 and Ps STUDY 2 Ps STUDY 1 Ps STUDY 2 STELARA ® STELARA ® Placebo 45 mg 90 mg Placebo 45 mg 90 mg Subjects randomized 255 255 256 410 409 411 PASI 75 response 8 (3%) 171 (67%) 170 (66%) 15 (4%) 273 (67%) 311 (76%) PGA of Cleared or Minimal 10 (4%) 151 (59%) 156 (61%) 18 (4%) 277 (68%) 300 (73%) Examination of age, gender, and race subgroups did not identify differences in response to STELARA ®among these subgroups.
In subjects who weighed 100 kg or less, response rates were comparable with both the 45 mg and 90 mg doses; however, in subjects who weighed greater than 100 kg, higher response rates were seen with 90 mg dosing compared with 45 mg dosing (Table 9 below).
Table 9: Clinical Outcomes by Weight at Week 12 in Adults with Plaque Psoriasis in Ps STUDY 1 and Ps STUDY 2 Ps STUDY 1 Ps STUDY 2 STELARA ® STELARA ® Placebo 45 mg 90 mg Placebo 45 mg 90 mg - *
- Patients were dosed with trial medication at Weeks 0 and 4.
Subjects randomized 255 255 256 410 409 411 PASI 75 response * ≤100 kg 4% 74% 65% 4% 73% 78% 6/166 124/168 107/164 12/290 218/297 225/289 >100 kg 2% 54% 68% 3% 49% 71% 2/89 47/87 63/92 3/120 55/112 86/121 PGA of Cleared or Minimal * ≤100 kg 4% 64% 63% 5% 74% 75% 7/166 108/168 103/164 14/290 220/297 216/289 >100 kg 3% 49% 58% 3% 51% 69% 3/89 43/87 53/92 4/120 57/112 84/121 Subjects in Ps STUDY 1 who were PASI 75 responders at both Weeks 28 and 40 were re-randomized at Week 40 to either continued dosing of STELARA ®(STELARA ®at Week 40) or to withdrawal of therapy (placebo at Week 40). At Week 52, 89% (144/162) of subjects re-randomized to STELARA ®treatment were PASI 75 responders compared with 63% (100/159) of subjects re-randomized to placebo (treatment withdrawal after Week 28 dose). The median time to loss of PASI 75 response among the subjects randomized to treatment withdrawal was 16 weeks.
14.2 Pediatric Plaque Psoriasis
A multicenter, randomized, double blind, placebo-controlled trial (Ps STUDY 3) enrolled 110 pediatric subjects 12 to 17 years of age with a minimum BSA involvement of 10%, a PASI score greater than or equal to 12, and a PGA score greater than or equal to 3, who were candidates for phototherapy or systemic therapy and whose disease was inadequately controlled by topical therapy.
Subjects were randomized to receive placebo (n = 37), the recommended dose of STELARA ®(n = 36), or one-half the recommended dose of STELARA ®(n = 37) by subcutaneous injection at Weeks 0 and 4 followed by dosing every 12 weeks (q12w). The recommended dose of STELARA ®was 0.75 mg/kg for subjects weighing less than 60 kg, 45 mg for subjects weighing 60 kg to 100 kg, and 90 mg for subjects weighing greater than 100 kg. At Week 12, subjects who received placebo were crossed over to receive STELARA ®at the recommended dose or one-half the recommended dose.
Of the pediatric subjects, approximately 63% had prior exposure to phototherapy or conventional systemic therapy and approximately 11% had prior exposure to biologics.
The endpoints were the proportion of subjects who achieved a PGA score of cleared (0) or minimal (1), PASI 75, and PASI 90 at Week 12. Subjects were followed for up to 60 weeks following first administration of trial agent.
Clinical Response
The efficacy results at Week 12 for Ps STUDY 3 are presented in Table 10.
Table 10: Efficacy Results at Week 12 in Pediatric Subjects 12 to 17 years with Plaque Psoriasis in Ps STUDY 3 Ps STUDY 3 Placebo
n (%)STELARA ®*
n (%)- *
- Using the weight-based dosage regimen specified in Table 1 and Table 2.
N 37 36 PGA PGA of cleared (0) or minimal (1) 2 (5.4%) 25 (69.4%) PASI PASI 75 responders 4 (10.8%) 29 (80.6%) PASI 90 responders 2 (5.4%) 22 (61.1%) 14.3 Psoriatic Arthritis
The safety and efficacy of STELARA ®was assessed in 927 patients (PsA STUDY 1, n=615; PsA STUDY 2, n=312), in two randomized, double-blind, placebo-controlled trials in adult patients 18 years of age and older with active PsA (≥5 swollen joints and ≥5 tender joints) despite nonsteroidal anti-inflammatory (NSAID) or disease modifying antirheumatic (DMARD) therapy. Patients in these trials had a diagnosis of PsA for at least 6 months. Patients with each subtype of PsA were enrolled, including polyarticular arthritis with the absence of rheumatoid nodules (39%), spondylitis with peripheral arthritis (28%), asymmetric peripheral arthritis (21%), distal interphalangeal involvement (12%) and arthritis mutilans (0.5%). Over 70% and 40% of the patients, respectively, had enthesitis and dactylitis at baseline.
Patients were randomized to receive treatment with STELARA ®45 mg, 90 mg, or placebo subcutaneously at Weeks 0 and 4 followed by every 12 weeks (q12w) dosing. Approximately 50% of patients continued on stable doses of MTX (≤25 mg/week). The primary endpoint was the percentage of patients achieving ACR 20 response at Week 24.
In PsA STUDY 1 and PsA STUDY 2, 80% and 86% of the patients, respectively, had been previously treated with DMARDs. In PsA STUDY 1, previous treatment with anti-tumor necrosis factor (TNF)-α agent was not allowed. In PsA STUDY 2, 58% (n=180) of the patients had been previously treated with TNF blocker, of whom over 70% had discontinued their TNF blocker treatment for lack of efficacy or intolerance at any time.
Clinical Response
In both trials, a greater proportion of patients achieved ACR 20, ACR 50 and PASI 75 response in the STELARA ®45 mg and 90 mg groups compared to placebo at Week 24 (see Table 11). ACR 70 responses were also higher in the STELARA ®45 mg and 90 mg groups, although the difference was only numerical (p=NS) in STUDY 2. Responses were consistent in patients treated with STELARA ®alone or in combination with methotrexate. Responses were similar in patients regardless of prior TNFα exposure.
Table 11: ACR 20, ACR 50, ACR 70 and PASI 75 responses in PsA STUDY 1 and PsA STUDY 2 at Week 24 PsA STUDY 1 PsA STUDY 2 STELARA ® STELARA ® Placebo 45 mg 90 mg Placebo 45 mg 90 mg - *
- Number of patients with ≥ 3% BSA psoriasis skin involvement at baseline
Number of patients randomized 206 205 204 104 103 105 ACR 20 response, N (%) 47 (23%) 87 (42%) 101 (50%) 21 (20%) 45 (44%) 46 (44%) ACR 50 response, N (%) 18 (9%) 51 (25%) 57 (28%) 7 (7%) 18 (17%) 24 (23%) ACR 70 response, N (%) 5 (2%) 25 (12%) 29 (14%) 3 (3%) 7 (7%) 9 (9%) Number of patients with ≥ 3% BSA * 146 145 149 80 80 81 PASI 75 response, N (%) 16 (11%) 83 (57%) 93 (62%) 4 (5%) 41 (51%) 45 (56%) The percent of patients achieving ACR 20 responses by visit is shown in Figure 1.
Figure 1: Percent of patients achieving ACR 20 response through Week 24 PsA STUDY 1 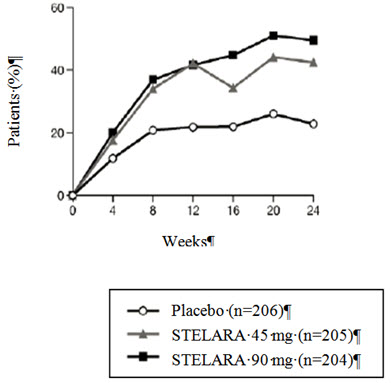
The results of the components of the ACR response criteria are shown in Table 12.
Table 12: Mean change from baseline in ACR components at Week 24 PsA STUDY 1 STELARA ® Placebo
(N = 206)45 mg
(N = 205)90 mg
(N = 204)- *
- Number of swollen joints counted (0–66)
- †
- Number of tender joints counted (0–68)
- ‡
- Visual analogue scale; 0= best, 10=worst.
- §
- Disability Index of the Health Assessment Questionnaire; 0 = best, 3 = worst, measures the patient's ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity.
- ¶
- CRP: (Normal Range 0.0–1.0 mg/dL)
Number of swollen joints * Baseline 15 12 13 Mean Change at Week 24 -3 -5 -6 Number of tender joints † Baseline 25 22 23 Mean Change at Week 24 -4 -8 -9 Patient's assessment of pain ‡ Baseline 6.1 6.2 6.6 Mean Change at Week 24 -0.5 -2.0 -2.6 Patient global assessment ‡ Baseline 6.1 6.3 6.4 Mean Change at Week 24 -0.5 -2.0 -2.5 Physician global assessment ‡ Baseline 5.8 5.7 6.1 Mean Change at Week 24 -1.4 -2.6 -3.1 Disability index (HAQ) § Baseline 1.2 1.2 1.2 Mean Change at Week 24 -0.1 -0.3 -0.4 CRP (mg/dL) ¶ Baseline 1.6 1.7 1.8 Mean Change at Week 24 0.01 -0.5 -0.8 An improvement in enthesitis and dactylitis scores was observed in each STELARA ®group compared with placebo at Week 24.
Physical Function
STELARA ®-treated patients showed improvement in physical function compared to patients treated with placebo as assessed by HAQ-DI at Week 24. In both trials, the proportion of HAQ-DI responders (≥0.3 improvement in HAQ-DI score) was greater in the STELARA ®45 mg and 90 mg groups compared to placebo at Week 24.
14.4 Crohn's Disease
STELARA ®was evaluated in three randomized, double-blind, placebo-controlled clinical trials in adult patients with moderately to severely active Crohn's disease (Crohn's Disease Activity Index [CDAI] score of 220 to 450). There were two 8-week intravenous induction trials(CD-1 and CD-2) followed by a 44-week subcutaneous randomized withdrawal maintenance trial (CD-3) representing 52 weeks of therapy. Patients in CD-1 had failed or were intolerant to treatment with one or more TNF blockers, while patients in CD-2 had failed or were intolerant to treatment with immunomodulators or corticosteroids, but never failed treatment with a TNF blocker.
Trials CD-1 and CD-2
In trials CD-1 and CD-2, 1409 patients were randomized, of whom 1368 (CD-1, n=741; CD-2, n=627) were included in the final efficacy analysis. Induction of clinical response (defined as a reduction in CDAI score of greater than or equal to 100 points or CDAI score of less than 150) at Week 6 and clinical remission (defined as a CDAI score of less than 150) at Week 8 were evaluated. In both trials, patients were randomized to receive a single intravenous administration of STELARA ®at either approximately 6 mg/kg, placebo (see Table 4), or 130 mg (a lower dose than recommended).
In trial CD-1, patients had failed or were intolerant to prior treatment with a TNF blocker: 29% patients had an inadequate initial response (primary non-responders), 69% responded but subsequently lost response (secondary non-responders) and 36% were intolerant to a TNF blocker. Of these patients, 48% failed or were intolerant to one TNF blocker and 52% had failed 2 or 3 prior TNF blockers. At baseline and throughout the trial, approximately 46% of the patients were receiving corticosteroids and 31% of the patients were receiving immunomodulators (AZA, 6-MP, MTX). The median baseline CDAI score was 319 in the STELARA ®approximately 6 mg/kg group and 313 in the placebo group.
In trial CD-2, patients had failed or were intolerant to prior treatment with corticosteroids (81% of patients), at least one immunomodulator (6-MP, AZA, MTX; 68% of patients), or both (49% of patients). Additionally, 69% never received a TNF blocker and 31% previously received but had not failed a TNF blocker. At baseline, and throughout the trial, approximately 39% of the patients were receiving corticosteroids and 35% of the patients were receiving immunomodulators (AZA, 6-MP, MTX). The median baseline CDAI score was 286 in the STELARA ®and 290 in the placebo group.
In these induction trials, a greater proportion of patients treated with STELARA ®(at the recommended dose of approximately 6 mg/kg dose) achieved clinical response at Week 6 and clinical remission at Week 8 compared to placebo (see Table 13for clinical response and remission rates). Clinical response and remission were significant as early as Week 3 in STELARA ®-treated patients and continued to improve through Week 8.
Table 13: Induction of Clinical Response and Remission in CD-1 *and CD-2 † CD-1
n = 741CD-2
n = 627Placebo
N = 247STELARA ®‡
N = 249Treatment difference and 95% CI Placebo
N = 209STELARA ®‡
N = 209Treatment difference and 95% CI Clinical remission is defined as CDAI score < 150; Clinical response is defined as reduction in CDAI score by at least 100 points or being in clinical remission: 70 point response is defined as reduction in CDAI score by at least 70 points - *
- Patient population consisted of patients who failed or were intolerant to TNF blocker therapy
- †
- Patient population consisted of patients who failed or were intolerant to corticosteroids or immunomodulators (e.g., 6-MP, AZA, MTX) and previously received but not failed a TNF blocker or were never treated with a TNF blocker.
- ‡
- Infusion dose of STELARA ® using the weight-based dosage regimen specified in Table 4.
- §
- 0.001≤ p < 0.01
- ¶
- p < 0.001
Clinical Response (100 point), Week 6 53 (21%) 84 (34%) § 12%
(4%, 20%)60 (29%) 116 (56%) ¶ 27%
(18%, 36%)Clinical Remission, Week 8 18 (7%) 52 (21%) ¶ 14%
(8%, 20%)41 (20%) 84 (40%) ¶ 21%
(12%, 29%)Clinical Response (100 point), Week 8 50 (20%) 94 (38%) ¶ 18%
(10%, 25%)67 (32%) 121 (58%) ¶ 26%
(17%, 35%)70 Point Response, Week 6 75 (30%) 109 (44%) § 13%
(5%, 22%)81 (39%) 135 (65%) ¶ 26%
(17%, 35%)70 Point Response, Week 3 67 (27%) 101 (41%) § 13%
(5%, 22%)66 (32%) 106 (51%) ¶ 19%
(10%, 28%)Trial CD-3
The maintenance trial (CD-3), evaluated 388 patients who achieved clinical response (≥100 point reduction in CDAI score) at Week 8 with either induction dose of STELARA ®in trials CD-1 or CD-2. Patients were randomized to receive a subcutaneous maintenance regimen of either 90 mg STELARA ®every 8 weeks or placebo for 44 weeks (see Table 14).
Table 14: Clinical Response and Remission in CD-3 (Week 44; 52 weeks from initiation of the induction dose) Placebo * 90 mg STELARA ®every 8 weeks Treatment difference and 95% CI N = 131 † N = 128 † Clinical remission is defined as CDAI score < 150; Clinical response is defined as reduction in CDAI of at least 100 points or being in clinical remission - *
- The placebo group consisted of patients who were in response to STELARA ® and were randomized to receive placebo at the start of maintenance therapy.
- †
- Patients who achieved clinical response to STELARA ® at the end of the induction trial.
- ‡
- p < 0.01
- §
- 0.01≤ p < 0.05
- ¶
- Patients in remission at the end of maintenance therapy who were in remission at the start of maintenance therapy. This does not account for any other time point during maintenance therapy.
Clinical Remission 47 (36%) 68 (53%) ‡ 17% (5%, 29%) Clinical Response 58 (44%) 76 (59%) § 15% (3%, 27%) Clinical Remission in patients in remission at the start of maintenance therapy ¶ 36/79 (46%) 52/78 (67%) ‡ 21% (6%, 36%) At Week 44, 47% of patients who received STELARA ®were corticosteroid-free and in clinical remission, compared to 30% of patients in the placebo group.
At Week 0 of trial CD-3, 34/56 (61%) STELARA ®-treated patients who previously failed or were intolerant to TNF blocker therapies were in clinical remission and 23/56 (41%) of these patients were in clinical remission at Week 44. In the placebo arm, 27/61 (44%) patients were in clinical remission at Week 0 while 16/61 (26%) of these patients were in remission at Week 44.
At Week 0 of trial CD-3, 46/72 (64%) STELARA ®-treated patients who had previously failed immunomodulator therapy or corticosteroids (but not TNF blockers) were in clinical remission and 45/72 (63%) of these patients were in clinical remission at Week 44. In the placebo arm, 50/70 (71%) of these patients were in clinical remission at Week 0 while 31/70 (44%) were in remission at Week 44. In the subset of these patients who were also naïve to TNF blockers, 34/52 (65%) of STELARA ®-treated patients were in clinical remission at Week 44 as compared to 25/51 (49%) in the placebo arm.
Patients who were not in clinical response 8 weeks after STELARA ®induction were not included in the primary efficacy analyses for trial CD-3; however, these patients were eligible to receive a 90 mg subcutaneous injection of STELARA ®upon entry into trial CD-3. Of these patients, 102/219 (47%) achieved clinical response eight weeks later and were followed for the duration of the trial.
14.5 Ulcerative Colitis
STELARA ®was evaluated in two randomized, double-blind, placebo-controlled clinical trials [UC-1 and UC-2 (NCT02407236)] in adult patients with moderately to severely active ulcerative colitis who had an inadequate response to or failed to tolerate a biologic (i.e., TNF blocker and/or vedolizumab), corticosteroids, and/or 6-MP or AZA therapy. The 8-week intravenous induction trial (UC-1) was followed by the 44-week subcutaneous randomized withdrawal maintenance trial (UC-2) for a total of 52 weeks of therapy.
Disease assessment was based on the Mayo score, which ranged from 0 to 12 and has four subscores that were each scored from 0 (normal) to 3 (most severe): stool frequency, rectal bleeding, findings on centrally-reviewed endoscopy, and physician global assessment. Moderately to severely active ulcerative colitis was defined at baseline (Week 0) as Mayo score of 6 to 12, including a Mayo endoscopy subscore ≥2. An endoscopy score of 2 was defined by marked erythema, absent vascular pattern, friability, erosions; and a score of 3 was defined by spontaneous bleeding, ulceration. At baseline, patients had a median Mayo score of 9, with 84% of patients having moderate disease (Mayo score 6–10) and 15% having severe disease (Mayo score 11–12).
Patients in these trials may have received other concomitant therapies including aminosalicylates, immunomodulatory agents (AZA, 6-MP, or MTX), and oral corticosteroids (prednisone).
Trial UC-1
In UC-1, 961 patients were randomized at Week 0 to a single intravenous administration of STELARA ®of approximately 6 mg/kg, 130 mg (a lower dose than recommended), or placebo. Patients enrolled in UC-1 had to have failed therapy with corticosteroids, immunomodulators or at least one biologic. A total of 51% had failed at least one biologic and 17% had failed both a TNF blocker and an integrin receptor blocker. Of the total population, 46% had failed corticosteroids or immunomodulators but were biologic-naïve and an additional 3% had previously received but had not failed a biologic. At induction baseline and throughout the trial, approximately 52% patients were receiving oral corticosteroids, 28% patients were receiving immunomodulators (AZA, 6-MP, or MTX) and 69% patients were receiving aminosalicylates.
The primary endpoint was clinical remission at Week 8. Clinical remission with a definition of: Mayo stool frequency subscore of 0 or 1, Mayo rectal bleeding subscore of 0 (no rectal bleeding), and Mayo endoscopy subscore of 0 or 1 (Mayo endoscopy subscore of 0 defined as normal or inactive disease and Mayo subscore of 1 defined as presence of erythema, decreased vascular pattern and no friability) is provided in Table 15.
The secondary endpoints were clinical response, endoscopic improvement, and histologic-endoscopic mucosal improvement. Clinical response with a definition of (≥ 2 points and ≥ 30% decrease in modified Mayo score, defined as 3-component Mayo score without the Physician's Global Assessment, with either a decrease from baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1), endoscopic improvement with a definition of Mayo endoscopy subscore of 0 or 1, and histologic-endoscopic mucosal improvement with a definition of combined endoscopic improvement and histologic improvement of the colon tissue [neutrophil infiltration in <5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue]) are provided in Table 15.
In UC-1, a significantly greater proportion of patients treated with STELARA ®(at the recommended dose of approximately 6 mg/kg dose) were in clinical remission and response and achieved endoscopic improvement and histologic-endoscopic mucosal improvement compared to placebo (see Table 15).
Table 15: Proportion of Patients Meeting Efficacy Endpoints at Week 8 in UC-1
EndpointPlacebo
N = 319STELARA ®*
N = 322Treatment difference and 97.5% CI † N % N % - *
- Infusion dose of STELARA ® using the weight-based dosage regimen specified in Table 4.
- †
- Adjusted treatment difference (97.5% CI)
- ‡
- Clinical remission was defined as Mayo stool frequency subscore of 0 or 1, Mayo rectal bleeding subscore of 0, and Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
- §
- p < 0.001
- ¶
- An additional 7 patients on placebo and 9 patients on STELARA ® (6 mg/kg) had been exposed to, but had not failed, biologics.
- #
- Endoscopic improvement was defined as Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
- Þ
- Clinical response was defined as a decrease from baseline in the modified Mayo score by ≥30% and ≥2 points, with either a decrease from baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1.
- ß
- Histologic-endoscopic mucosal improvement was defined as combined endoscopic improvement (Mayo endoscopy subscore of 0 or 1) and histologic improvement of the colon tissue (neutrophil infiltration in <5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue).
Clinical Remission ‡ 22 7% 62 19% 12%
(7%, 18%) §Bio-naïve ¶ 14/151 9% 36/147 24% Prior biologic failure 7/161 4% 24/166 14% Endoscopic Improvement # 40 13% 80 25% 12%
(6%, 19%) §Bio-naïve ¶ 28/151 19% 43/147 29% Prior biologic failure 11/161 7% 34/166 20% Clinical Response Þ 99 31% 186 58% 27%
(18%, 35%) §Bio-naïve ¶ 55/151 36% 94/147 64% Prior biologic failure 42/161 26% 86/166 52% Histologic-Endoscopic Mucosal Improvement ß 26 8% 54 17% 9%
(3%, 14%) §Bio-naïve ¶ 19/151 13% 30/147 20% Prior biologic failure 6/161 4% 21/166 13% The relationship of histologic-endoscopic mucosal improvement, as defined in UC-1, at Week 8 to disease progression and long-term outcomes was not evaluated during UC-1.
Trial UC-2
The maintenance trial (UC-2) evaluated 523 patients who achieved clinical response 8 weeks following the intravenous administration of either induction dose of STELARA ®in UC-1. These patients were randomized to receive a subcutaneous maintenance regimen of either 90 mg STELARA ®every 8 weeks, or every 12 weeks (a lower dose than recommended), or placebo for 44 weeks.
The primary endpoint was the proportion of patients in clinical remission at Week 44. The secondary endpoints included the proportion of patients maintaining clinical response at Week 44, the proportion of patients with endoscopic improvement at Week 44, the proportion of patients with corticosteroid-free clinical remission at Week 44, and the proportion of patients maintaining clinical remission at Week 44 among patients who achieved clinical remission 8 weeks after induction.
Results of the primary and secondary endpoints at Week 44 in patients treated with STELARA ®at the recommended dosage (90 mg every 8 weeks) compared to the placebo are shown in Table 16.
Table 16: Efficacy Endpoints of Maintenance at Week 44 in UC-2 (52 Weeks from Initiation of the Induction Dose) Endpoint Placebo *
N = 175 †90 mg STELARA ®every 8 weeks
N = 176Treatment difference and 95% CI N % N % - *
- The placebo group consisted of patients who were in response to STELARA ® and were randomized to receive placebo at the start of maintenance therapy.
- †
- Clinical response was defined as a decrease from baseline in the modified Mayo score by ≥30% and ≥2 points, with either a decrease from baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1.
- ‡
- Clinical remission was defined as Mayo stool frequency subscore of 0 or 1, Mayo rectal bleeding subscore of 0, and Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
- §
- p =<0.001
- ¶
- An additional 3 patients on placebo and 6 patients on STELARA ® had been exposed to, but had not failed, biologics.
- #
- Endoscopic improvement was defined as Mayo endoscopy subscore of 0 or 1 (modified so that 1 does not include friability).
- Þ
- Corticosteroid-free clinical remission was defined as patients in clinical remission and not receiving corticosteroids at Week 44.
- ß
- p=0.004
Clinical Remission ‡ 46 26% 79 45% 19%
(9%, 28%) §Bio-naïve ¶ 30/84 36% 39/79 49% Prior biologic failure 16/88 18% 37/91 41% Maintenance of Clinical Response at Week 44 † 84 48% 130 74% 26%
(16%, 36%) §Bio-naïve ¶ 49/84 58% 62/79 78% Prior biologic failure 35/88 40% 64/91 70% Endoscopic Improvement # 47 27% 83 47% 20%
(11%, 30%) §Bio-naïve ¶ 29/84 35% 42/79 53% Prior biologic failure 18/88 20% 38/91 42% Corticosteroid-free Clinical Remission Þ 45 26% 76 43% 17%
(8%, 27%) §Bio-naïve ¶ 30/84 36% 38/79 48% Prior biologic failure 15/88 17% 35/91 38% Maintenance of Clinical Remission at Week 44 in patients who achieved clinical remission 8 weeks after induction 18/50 36% 27/41 66% 31%
(12%, 50%) ßBio-naïve ¶ 12/27 44% 14/20 70% Prior biologic failure 6/23 26% 12/18 67% Other Endpoints
Week 16 Responders to Ustekinumab Induction
Patients who were not in clinical response 8 weeks after induction with STELARA ®in UC-1 were not included in the primary efficacy analyses for trial UC-2; however, these patients were eligible to receive a 90 mg subcutaneous injection of STELARA ®at Week 8. Of these patients, 55/101 (54%) achieved clinical response eight weeks later (Week 16) and received STELARA ®90 mg subcutaneously every 8 weeks during the UC-2 trial. At Week 44, there were 97/157 (62%) patients who maintained clinical response and there were 51/157 (32%) who achieved clinical remission.
Histologic-Endoscopic Mucosal Improvement at Week 44
The proportion of patients achieving histologic-endoscopic mucosal improvement during maintenance treatment in UC-2 was 75/172 (44%) among patients on STELARA ®and 40/172 (23%) in patients on placebo at Week 44. The relationship of histologic-endoscopic mucosal improvement, as defined in UC-2, at Week 44 to progression of disease or long-term outcomes was not evaluated in UC-2.
Endoscopic Normalization
Normalization of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0. At Week 8 in UC-1, endoscopic normalization was achieved in 25/322 (8%) of patients treated with STELARA ®and 12/319 (4%) of patients in the placebo group. At Week 44 of UC-2, endoscopic normalization was achieved in 51/176 (29%) of patients treated with STELARA ®and in 32/175 (18%) of patients in placebo group.
-
15 REFERENCES
- Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 6.6.2 Regs Research Data, Nov 2009 Sub (1973–2007) - Linked To County Attributes - Total U.S., 1969–2007 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2010, based on the November 2009 submission.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
STELARA ®(ustekinumab) injection is a sterile, preservative-free, colorless to light yellow solution and may contain a few small translucent or white particles. It is supplied as individually packaged, single-dose prefilled syringes or single-dose vials.
For Subcutaneous Use
Prefilled Syringes
- 45 mg/0.5 mL (NDC 57894-060-03)
- 90 mg/mL (NDC 57894-061-03)
Each prefilled syringe is equipped with a 27 -gauge fixed ½ inch needle, a needle safety guard, and a needle cover that contains dry natural rubber.
Single-dose Vial
- 45 mg/0.5 mL (NDC 57894-060-02)
Storage and Stability
Store STELARA ®vials and prefilled syringes refrigerated between 2°C to 8°C (36°F to 46°F). Store STELARA ®vials upright. Keep the product in the original carton to protect from light until the time of use. Do not freeze. Do not shake.
If needed, individual prefilled syringes may be stored at room temperature up to 30°C (86°F) for a maximum single period of up to 30 days in the original carton to protect from light. Record the date when the prefilled syringe is first removed from the refrigerator on the carton in the space provided. Once a syringe has been stored at room temperature, do not return to the refrigerator. Discard the syringe if not used within 30 days at room temperature storage. Do not use STELARA ®after the expiration date on the carton or on the prefilled syringe.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Infections
Inform patients that STELARA ®may lower the ability of their immune system to fight infections and to contact their healthcare provider immediately if they develop any signs or symptoms of infection [see Warnings and Precautions (5.1)] .
Malignancies
Inform patients of the risk of developing malignancies while receiving STELARA ®[see Warnings and Precautions (5.4)] .
Hypersensitivity Reactions
- Advise patients to seek immediate medical attention if they experience any signs or symptoms of serious hypersensitivity reactions and discontinue STELARA ®[see Warnings and Precautions (5.5)].
- Inform patients the needle cover on the prefilled syringe contains dry natural rubber (a derivative of latex), which may cause allergic reactions in individuals sensitive to latex [see Dosage and Administration (2.4)].
Posterior Reversible Encephalopathy Syndrome (PRES)
Inform patients to immediately contact their healthcare provider if they experience signs and symptoms of PRES (which may include headache, seizures, confusion, or visual disturbances) [see Warnings and Precautions (5.6)] .
Immunizations
Inform patients that STELARA ®can interfere with the usual response to immunizations and that they should avoid live vaccines [see Warnings and Precautions (5.7)] .
-
SPL UNCLASSIFIED SECTION
Prefilled Syringe Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, US License No. 1864 at Baxter Pharmaceutical Solutions, Bloomington, IN 47403 and at Cilag AG, Schaffhausen, Switzerland
Vial Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, US License No. 1864 at Cilag AG, Schaffhausen, Switzerland
© 2012, 2016, 2019 Janssen Pharmaceutical Companies
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: MM/YYYY MEDICATION GUIDE
STELARA ®(stel ar' a)
(ustekinumab)
injection, for subcutaneous or intravenous useWhat is the most important information I should know about STELARA?
STELARA is a medicine that affects your immune system. STELARA can increase your risk of having serious side effects, including:
Serious infections.STELARA may lower the ability of your immune system to fight infections and may increase your risk of infections. Some people have serious infections while taking STELARA, including tuberculosis (TB), and infections caused by bacteria, fungi, or viruses. Some people have to be hospitalized for treatment of their infection.- Your doctor should check you for TB before starting STELARA.
- If your doctor feels that you are at risk for TB, you may be treated with medicine for TB before you begin treatment with STELARA and during treatment with STELARA.
- Your doctor should watch you closely for signs and symptoms of TB while you are being treated with STELARA.
You should not start taking STELARA if you have any kind of infection unless your doctor says it is okay.
Before starting STELARA, tell your doctor if you:
- think you have an infection or have symptoms of an infection such as:
- fever, sweat, or chills
- muscle aches
- cough
- shortness of breath
- blood in phlegm
- weight loss
- warm, red, or painful skin or sores on your body
- diarrhea or stomach pain
- burning when you urinate or urinate more often than normal
- feel very tired
- are being treated for an infection or have any open cuts.
- get a lot of infections or have infections that keep coming back.
- have TB, or have been in close contact with someone with TB.
After starting STELARA, call your doctor right away if you have any symptoms of an infection (see above). These may be signs of infections such as chest infections, or skin infections or shingles that could have serious complications. STELARA can make you more likely to get infections or make an infection that you have worse.
People who have a genetic problem where the body does not make any of the proteins interleukin 12 (IL-12) and interleukin 23 (IL-23) are at a higher risk for certain serious infections. These infections can spread throughout the body and cause death. People who take STELARA may also be more likely to get these infections.
Cancers.STELARA may decrease the activity of your immune system and increase your risk for certain types of cancers. Tell your doctor if you have ever had any type of cancer. Some people who are receiving STELARA and have risk factors for skin cancer have developed certain types of skin cancers. During your treatment with STELARA, tell your doctor if you develop any new skin growths.
Posterior Reversible Encephalopathy Syndrome (PRES).PRES is a rare condition that affects the brain and can cause death. The cause of PRES is not known. If PRES is found early and treated, most people recover. Tell your doctor right away if you have any new or worsening medical problems including:
- headache
- seizures
- confusion
- vision problems
What is STELARA?
STELARA is a prescription medicine used to treat:- adults and children 6 years and older with moderate or severe psoriasis who may benefit from taking injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light alone or with pills).
- adults and children 6 years and older with active psoriatic arthritis.
- adults 18 years and older with moderately to severely active Crohn's disease.
- adults 18 years and older with moderately to severely active ulcerative colitis.
It is not known if STELARA is safe and effective in children less than 6 years of age.
Do not take STELARA if you areallergic to ustekinumab or any of the ingredients in STELARA. See the end of this Medication Guide for a complete list of ingredients in STELARA. Before you receive STELARA, tell your doctor about all of your medical conditions, including if you:
- have any of the conditions or symptoms listed in the section " What is the most important information I should know about STELARA?"
- ever had an allergic reaction to STELARA. Ask your doctor if you are not sure.
- are allergic to latex. The needle cover on the prefilled syringe contains latex.
- have recently received or are scheduled to receive an immunization (vaccine). People who take STELARA should not receive live vaccines. Tell your doctor if anyone in your house needs a live vaccine. The viruses used in some types of live vaccines can spread to people with a weakened immune system, and can cause serious problems. You should not receive the BCG vaccine during the one year before receiving STELARA or one year after you stop receiving STELARA.
- have any new or changing lesions within psoriasis areas or on normal skin.
- are receiving or have received allergy shots, especially for serious allergic reactions. Allergy shots may not work as well for you during treatment with STELARA. STELARA may also increase your risk of having an allergic reaction to an allergy shot.
- receive or have received phototherapy for your psoriasis.
- are pregnant or plan to become pregnant. It is not known if STELARA can harm your unborn baby. You and your doctor should decide if you will receive STELARA. See " What should I avoid while using STELARA?"
- received STELARA while you were pregnant. It is important that you tell your baby's healthcare provider before any vaccinations are given to your baby.
- are breastfeeding or plan to breastfeed. STELARA can pass into your breast milk
- Talk to your doctor about the best way to feed your baby if you receive STELARA.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I use STELARA? - Use STELARA exactly as your doctor tells you to.
- The needle cover on the STELARA prefilled syringe contains latex. Do not handle the needle cover if you are sensitive to latex.
- Adults with Crohn's disease and ulcerative colitis will receive the first dose of STELARA through a vein in the arm (intravenous infusion) in a healthcare facility by a healthcare provider. It takes at least 1 hour to receive the full dose of medicine. You will then receive STELARA as an injection under the skin (subcutaneous injection) 8 weeks after the first dose of STELARA, as described below.
- Adults with psoriasis or psoriatic arthritis, and children 6 years and older with psoriasis or psoriatic arthritis will receive STELARA as an injection under the skin (subcutaneous injection) as described below.
-
Injecting STELARA under your skin
- STELARA is intended for use under the guidance and supervision of your doctor. In children 6 years and older, it is recommended that STELARA be administered by a healthcare provider. If your doctor decides that you or a caregiver may give your injections of STELARA at home, you should receive training on the right way to prepare and inject STELARA. Your doctor will determine the right dose of STELARA for you, the amount for each injection, and how often you should receive it. Do not try to inject STELARA yourself until you or your caregiver have been shown how to inject STELARA by your doctor or nurse.
- Inject STELARA under the skin (subcutaneous injection) in your upper arms, buttocks, upper legs (thighs) or stomach area (abdomen).
- Do not give an injection in an area of the skin that is tender, bruised, red or hard.
- Use a different injection site each time you use STELARA.
- If you inject more STELARA than prescribed, call your doctor right away.
- Be sure to keep all of your scheduled follow-up appointments.
Read the detailed Instructions for Use at the end of this Medication Guide for instructions about how to prepare and inject a dose of STELARA, and how to properly throw away (dispose of) used needles and syringes. The syringe, needle and vial must never be re-used. After the rubber stopper is punctured, STELARA can become contaminated by harmful bacteria which could cause an infection if re-used. Therefore, throw away any unused portion of STELARA.
What should I avoid while using STELARA?
You should not receive a live vaccine while taking STELARA. See " Before you receive STELARA, tell your doctor about all of your medical conditions, including if you:"
What are the possible side effects of STELARA?
STELARA may cause serious side effects, including:- See " What is the most important information I should know about STELARA?"
- Serious allergic reactions.Serious allergic reactions can occur with STELARA. Stop using STELARA and get medical help right away if you have any of the following symptoms of a serious allergic reaction:
- feeling faint
- swelling of your face, eyelids, tongue, or throat
- chest tightness
- skin rash
- Lung inflammation.Cases of lung inflammation have happened in some people who receive STELARA, and may be serious. These lung problems may need to be treated in a hospital. Tell your doctor right away if you develop shortness of breath or a cough that doesn't go away during treatment with STELARA .
Common side effects of STELARA include: - nasal congestion, sore throat, and runny nose
- upper respiratory infections
- fever
- headache
- tiredness
- itching
- nausea and vomiting
- redness at the injection site
- vaginal yeast infections
- urinary tract infections
- sinus infection
- bronchitis
- diarrhea
- stomach pain
These are not all of the possible side effects of STELARA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Janssen Biotech, Inc. at 1-800 JANSSEN (1-800-526-7736).How should I store STELARA? - Store STELARA vials and prefilled syringes in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Store STELARA vials standing up straight.
- Store STELARA in the original carton to protect it from light until time to use it.
- Do not freeze STELARA.
- Do not shake STELARA.
If needed, individual STELARA prefilled syringes may also be stored at room temperature up to 30°C (86ºF) for a maximum single period of up to 30 days in the original carton to protect from light. Record the date when the prefilled syringe is first removed from the refrigerator on the carton in the space provided. Once a syringe has been stored at room temperature, it should not be returned to the refrigerator. Discard the syringe if not used within 30 days at room temperature storage. Do not use STELARA after the expiration date on the carton or on the prefilled syringe.
Keep STELARA and all medicines out of the reach of children.
General information about the safe and effective use of STELARA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use STELARA for a condition for which it was not prescribed. Do not give STELARA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about STELARA that was written for health professionals.What are the ingredients in STELARA?
Active ingredient: ustekinumab
Inactive ingredients: Single-dose prefilled syringe for subcutaneous usecontains L-histidine, L-histidine monohydrochloride monohydrate, Polysorbate 80, and sucrose. Single-dose vial for subcutaneous usecontains L-histidine, L-histidine hydrochloride monohydrate, Polysorbate 80 and sucrose. Single-dose vial for intravenous infusioncontains EDTA disodium salt dihydrate, L-histidine, L-histidine hydrochloride monohydrate, L-methionine, Polysorbate 80, and sucrose.
Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, US License No. 1864
© 2012, 2016, 2019 Janssen Pharmaceutical Companies
For more information, go to www.stelarainfo.comor call 1-800-JANSSEN (1-800-526-7736). -
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
STELARA ( stel ar' a )
(ustekinumab)
injection, for subcutaneous useInstructions for injecting STELARA using a prefilled syringe.
Read this Instructions for Use before you start using STELARA. Your doctor or nurse should show you how to prepare and give your injection of STELARA the right way.
If you cannot give yourself the injection:
- ask your doctor or nurse to help you, or
- ask someone who has been trained by a doctor or nurse to give your injections.
Do not try to inject STELARA yourself until you have been shown how to inject STELARA by your doctor, nurse or health professional.
Important information:
- Before you start, check the carton to make sure that it is the right dose. You will have either 45 mg or 90 mg as prescribed by your doctor.
- If your dose is 45 mg, you will receive one 45 mg prefilled syringe.
- If your dose is 90 mg, you will receive either one 90 mg prefilled syringe or two 45 mg prefilled syringes. If you receive two 45 mg prefilled syringes for a 90 mg dose, you will need to give yourself two injections, one right after the other.
- Children 12 years of age and older with psoriasis who weigh 132 pounds or more may use a prefilled syringe.
- Check the expiration date on the prefilled syringe and carton. If the expiration date has passed or if the prefilled syringe has been kept at room temperature up to 30ºC (86ºF) for longer than a maximum single period of 30 days or if the prefilled syringe has been stored above 30ºC (86ºF), do not use it. If the expiration date has passed or if the prefilled syringe has been stored above 30ºC (86ºF), call your doctor or pharmacist, or call 1-800-JANSSEN(1-800-526-7736) for help.
- Make sure the syringe is not damaged.
- The needle cover on the prefilled syringe contains latex. Do not handle the needle cover on the STELARA prefilled syringe if you are allergic to latex.
- Check your prefilled syringe for any particles or discoloration. Your prefilled syringe should look clear and colorless to light yellow with few white particles.
- Do not use if it is frozen, discolored, cloudy or has large particles. Get a new prefilled syringe.
- Do not shake the prefilled syringe at any time.Shaking your prefilled syringe may damage your STELARA medicine. If your prefilled syringe has been shaken, do not use it. Get a new prefilled syringe.
- To reduce the risk of accidental needle sticks, each prefilled syringe has a needle guard that is automatically activated to cover the needle after you have given your injection. Do not pull back on the plunger at any time.
Gather the supplies you will need to prepare and to give your injection. (See Figure A)
You will need:
- antiseptic wipes
- cotton balls or gauze pads
- adhesive bandage
- your prescribed dose of STELARA (See Figure B)
- FDA-cleared sharps disposal container. See " Step 4: Dispose of the syringe."
To prevent early activation of the needle safety guard, do not touch the NEEDLE GUARD ACTIVATION CLIPS at any time during use.
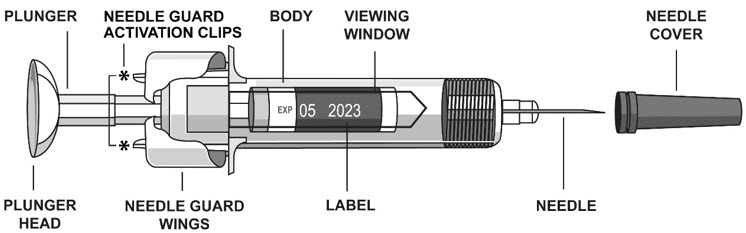
Step 1: Prepare the injection.
- Choose a well-lit, clean, flat work surface.
- Wash your hands well with soap and warm water.
- Hold the prefilled syringe with the covered needle pointing upward.
Step 2: Prepare your injection site
- Choose an injection site around your stomach area (abdomen), buttocks, upper legs (thighs). If a caregiver is giving you the injection, the outer area of the upper arms may also be used. (See Figure C)
- Use a different injection site for each injection. Do notgive an injection in an area of the skin that is tender, bruised, red or hard.
- Clean the skin with an antiseptic wipe where you plan to give your injection.
- Do nottouch this area again before giving the injection. Let your skin dry before injecting.
- Do notfan or blow on the clean area.
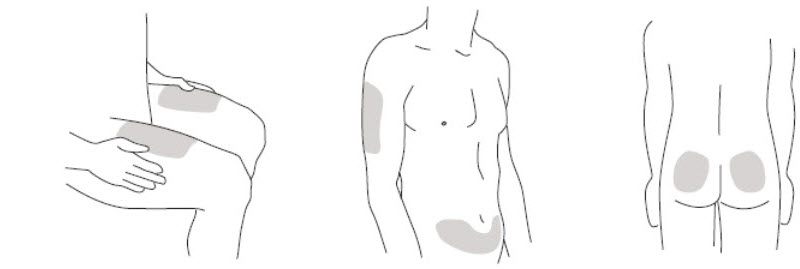
*Areas in gray are recommended injection sites.
Step 3: Inject STELARA
- Remove the needle cover when you are ready to inject your STELARA.
- Do nottouch the plunger while removing the needle cover.
- Hold the body of the prefilled syringe with one hand, and pull the needle cover straight off. (see Figure D)
- Put the needle cover in the trash.
- You may also see a drop of liquid at the end of the needle. This is normal.
- Do nottouch the needle or let it touch anything.
- Do notuse the prefilled syringe if it is dropped without the needle cover in place. Call your doctor, nurse or health professional for instructions.
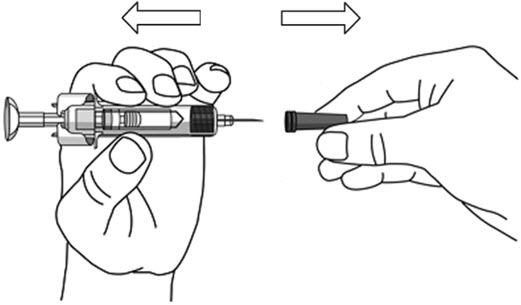
- Hold the body of the prefilled syringe in one hand between the thumb and index fingers. (See Figure E)
- Do notpull back on the plunger at any time.
- Use the other hand to gently pinch the cleaned area of skin. Hold firmly.
- Use a quick, dart-like motion to insert the needle into the pinched skin at about a 45-degree angle. (See Figure F)
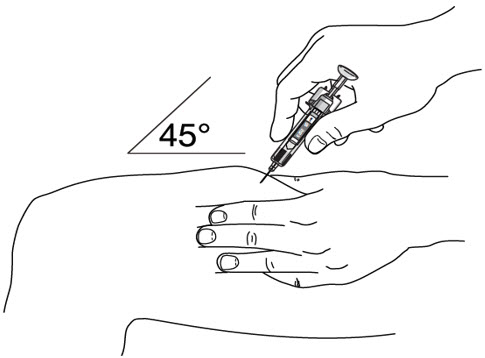
- Inject all of the liquid by using your thumb to push in the plunger until the plunger head is completely between the needle guard wings. (See Figure G)
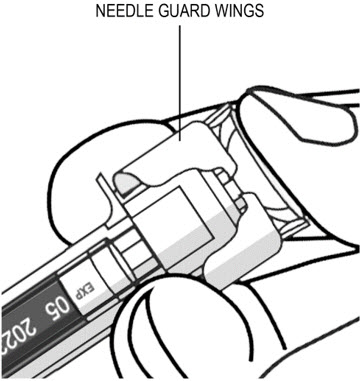
- When the plunger is pushed as far as it will go, keep pressure on the plunger head. Take the needle out of the skin and let go of the skin.
- Slowly take your thumb off the plunger head. This will let the empty syringe move up until the entire needle is covered by the needle guard. (See Figure H)
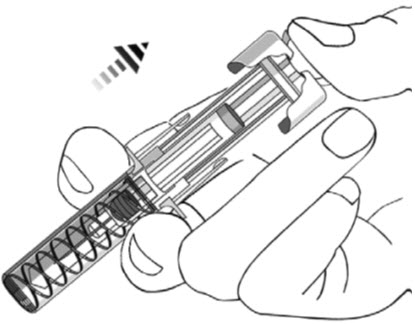
- When the needle is pulled out of your skin, there may be a little bleeding at the injection site. This is normal. You can press a cotton ball or gauze pad to the injection site if needed. Do not rub the injection site. You may cover the injection site with a small adhesive bandage, if necessary.
If your dose is 90 mg, you will receive either one 90 mg prefilled syringe or two 45 mg prefilled syringes. If you receive two 45 mg prefilled syringes for a 90 mg dose, you will need to give yourself a second injection right after the first. Repeat Steps 1–3 for the second injection using a new syringe. Choose a different site for the second injection.
Step 4: Dispose of the syringe.
- Put the syringe in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic.
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out.
- upright and stable during use,
- leak-resistant,
- and properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be local or state laws about how to throw away syringes and needles. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your sharps disposal container.
- If you have any questions, talk to your doctor or pharmacist.
Keep STELARA and all medicines out of the reach of children.
Prefilled Syringe Manufactured by:
Janssen Biotech, Inc., Horsham, PA 19044, US License No. 1864 at Baxter Pharmaceutical Solutions, Bloomington, IN 47403 and at Cilag AG, Schaffhausen, SwitzerlandThis Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 03/2020
© 2012 Janssen Pharmaceutical Companies
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
STELARA ( stel ar' a )
(ustekinumab)
injection, for subcutaneous useInstructions for injecting STELARA from a vial.
Read this Instructions for Use before you start using STELARA. Your doctor or nurse should show you how to prepare, measure your dose, and give your injection of STELARA the right way.
If you cannot give yourself the injection:
- ask your doctor or nurse to help you, or
- ask someone who has been trained by a doctor or nurse to give your injections.
Do not try to inject STELARA yourself until you have been shown how to inject STELARA by your doctor, nurse or health professional.
Important information:
- Before you start, check the carton to make sure that it is the right dose. You will have either 45 mg or 90 mg as prescribed by your doctor.
- If your dose is 45 mg or less you will receive one 45 mg vial.
- If your dose is 90 mg, you will receive two 45 mg vials and you will need to give yourself two injections, one right after the other.
- Children 12 years of age and older weighing less than 132 pounds require a dose lower than 45 mg.
- Check the expiration date on the vial and carton. If the expiration date has passed, do not use it. If the expiration date has passed, call your doctor or pharmacist, or call 1-800-JANSSEN (1-800-526-7736) for help.
- Check the vial for any particles or discoloration. Your vial should look clear and colorless to light yellow with few white particles.
- Do not use if it is frozen, discolored, cloudy or has large particles. Get a new vial.
- Do not shake the vial at any time.Shaking your vial may damage your STELARA medicine. If your vial has been shaken, do not use it. Get a new vial.
- Do not use a STELARA vial more than one time, even if there is medicine left in the vial. After the rubber stopper is punctured, STELARA can become contaminated by harmful bacteria which could cause an infection if re-used. Therefore, throw away any unused STELARA after you give your injection.
- Safely throw away (dispose of) STELARA vials after use.
- Do not re-use syringes or needles. See " Step 6: Dispose of needles and syringes."
- To avoid needle-stick injuries, do notrecap needles.
Gather the supplies you will need to prepare STELARA and to give your injection. (See Figure A)
You will need:
- a syringe with the needle attached, you will need a prescription from your healthcare provider to get syringes with the needles attached from your pharmacy.
- antiseptic wipes
- cotton balls or gauze pads
- adhesive bandage
- your prescribed dose of STELARA
- FDA-cleared sharps disposal container. See " Step 6: Dispose of needles and syringes."
Figure A
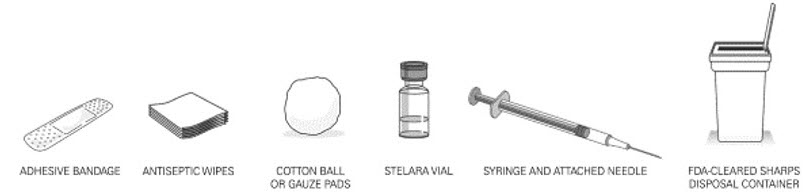
Step 1: Prepare the injection.
- Choose a well-lit, clean, flat work surface.
- Wash your hands well with soap and warm water.
Step 2: Prepare your injection site
- Choose an injection site around your stomach area (abdomen), buttocks, and upper legs (thighs).
If a caregiver is giving you the injection, the outer area of the upper arms may also be used. (See Figure B) - Use a different injection site for each injection. Do notgive an injection in an area of the skin that is tender, bruised, red or hard.
- Clean the skin with an antiseptic wipe where you plan to give your injection.
- Do nottouch this area again before giving the injection. Let your skin dry before injecting.
- Do notfan or blow on the clean area.
Figure B
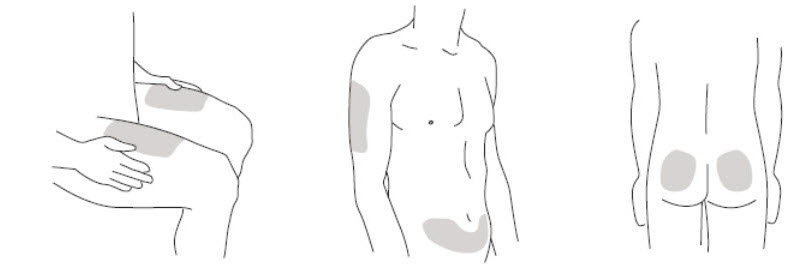
*Areas in gray are recommended injection sites.
Step 3: Prepare the vial.
- Remove the cap from the top of the vial. Throw away the cap but do not remove the rubber stopper. (See Figure C)
Figure C

- Clean the rubber stopper with an antiseptic swab. (See Figure D)
Figure D
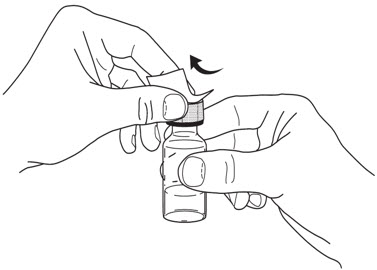
- Do not touch the rubber stopper after you clean it.
- Put the vial on a flat surface.
Step 4: Prepare the needle
- Pick up the syringe with the needle attached.
- Remove the cap that covers the needle. (See Figure E)
- Throw the needle cap away. Do not touch the needle or allow the needle to touch anything.
Figure E
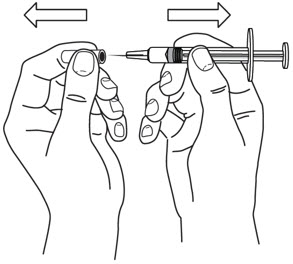
- Carefully pull back on the plunger to the line that matches the dose prescribed by your doctor.
- Hold the vial between your thumb and index (pointer) finger.
- Use your other hand to push the syringe needle through the center of the rubber stopper. (See Figure F)
Figure F
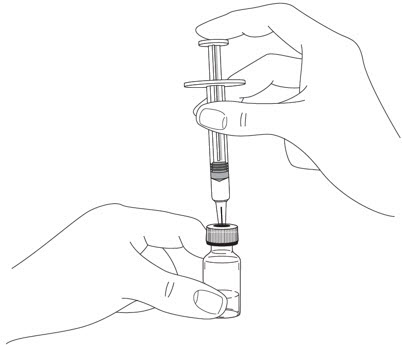
- Push down on the plunger until all of the air has gone from the syringe into the vial.
- Turn the vial and the syringe upside down. (See Figure G)
- Hold the STELARA vial with one hand.
- It is important that the needle is always in the liquid in order to prevent air bubbles forming in the syringe.
- Pull back on the syringe plunger with your other hand.
- Fill the syringe until the black tip of the plunger lines up with the mark that matches your prescribed dose.
Figure G
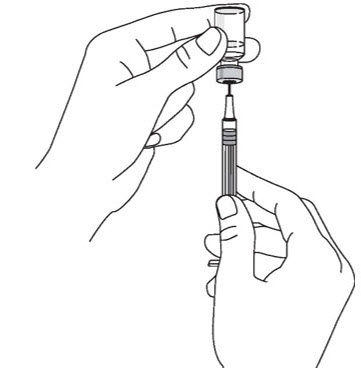
- Do not remove the needle from the vial.Hold the syringe with the needle pointing up to see if it has any air bubbles inside.
- If there are air bubbles, gently tap the side of the syringe until the air bubbles rise to the top. (See Figure H)
- Slowly press the plunger up until all of the air bubbles are out of the syringe (but none of the liquid is out).
- Remove the syringe from the vial. Do not lay the syringe down or allow the needle to touch anything.
Figure H
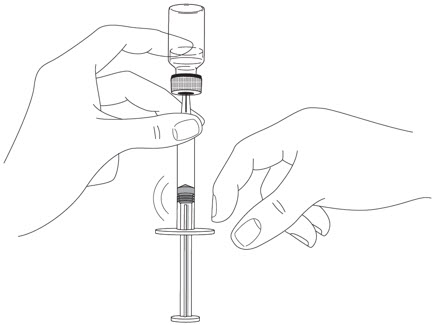
Step 5: Inject STELARA
- Hold the barrel of the syringe in one hand, between the thumb and index fingers.
- Do notpull back on the plunger at any time.
- Use the other hand to gently pinch the cleaned area of skin. Hold firmly.
- Use a quick, dart-like motion to insert the needle into the pinched skin at about a 45-degree angle. (See Figure I)
Figure I
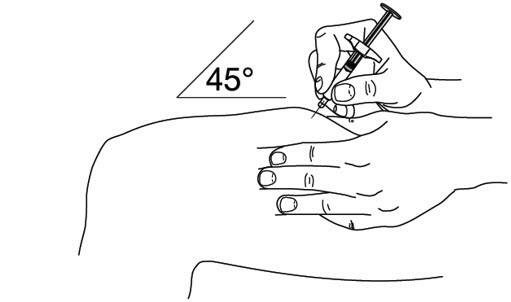
- Push the plunger with your thumb as far as it will go to inject all of the liquid. Push it slowly and evenly, keeping the skin gently pinched.
- When the syringe is empty, pull the needle out of your skin and let go of the skin. (See Figure J)
Figure J
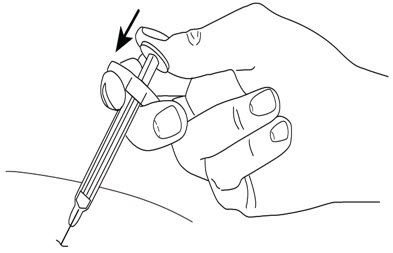
- When the needle is pulled out of your skin, there may be a little bleeding at the injection site. This is normal. You can press a cotton ball or gauze pad to the injection site if needed. Do not rub the injection site. You may cover the injection site with a small adhesive bandage, if necessary.
If your dose is 90 mg, you will receive two 45 mg vials and you will need to give yourself a second injection right after the first. Repeat Steps 1–5 using a new syringe.Choose a different site for the second injection.
Step 6: Dispose of the needles and syringes.
- Do notre-use a syringe or needle.
- To avoid needle-stick injuries, do not recap a needle.
- Put your needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant,
- and properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be local or state laws about how to throw away syringes and needles. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your sharps disposal container.
- Throw away the vial into the container where you put the syringes and needles.
- If you have any questions, talk to your doctor or pharmacist.
Keep STELARA and all medicines out of the reach of children.
Vial Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, US License No. 1864 at Cilag AG, Schaffhausen, Switzerland
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 03/2020
© 2012, 2016 Janssen Pharmaceutical Companies
-
PRINCIPAL DISPLAY PANEL - 45 mg/0.5 mL Vial Carton
Stelara®
(ustekinumab)
Injection45 mg/0.5 mL
For subcutaneous use
Contains one 45 mg/0.5 mL syringe
NDC 57894-060-03
Single-dose prefilled syringe - Discard unused portion
The 45 mg prefilled syringe contains:
45 mg ustekinumab, 0.5 mg L-histidine and L-histidine
monohydrochloride monohydrate, 0.02 mg polysorbate 80,
and 38 mg sucrose to fill a final volume of 0.5 mLSee package insert for dosing information
Rx only
ATTENTION: Dispense the enclosed Medication Guide
to each patient.© 2009 Janssen

-
PRINCIPAL DISPLAY PANEL - 90 mg/mL Syringe Carton
Stelara®
(ustekinumab)
Injection90 mg/mL
For subcutaneous use
Contains one 90 mg/mL syringe
NDC 57894-061-03
Single-dose prefilled syringe - Discard unused portion
The 90 mg prefilled syringe contains:
90 mg ustekinumab, 1 mg L-histidine and L-histidine
monohydrochloride monohydrate, 0.04 mg polysorbate 80,
and 76 mg sucrose to fill a final volume of 1 mLSee package insert for dosing information
Rx only
ATTENTION: Dispense the enclosed Medication
Guide to each patient.© 2009 Janssen
- PRINCIPAL DISPLAY PANEL - 130 mg/26 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
STELARA
ustekinumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-060 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength USTEKINUMAB (UNII: FU77B4U5Z0) (USTEKINUMAB - UNII:FU77B4U5Z0) USTEKINUMAB 45 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 38 mg in 0.5 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.02 mg in 0.5 mL HISTIDINE (UNII: 4QD397987E) 0.5 mg in 0.5 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-060-02 1 in 1 CARTON 09/25/2009 1 0.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:57894-060-03 1 in 1 CARTON 09/25/2009 2 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC:57894-060-04 1 in 1 CARTON 09/25/2009 3 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125261 09/25/2009 STELARA
ustekinumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-061 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength USTEKINUMAB (UNII: FU77B4U5Z0) (USTEKINUMAB - UNII:FU77B4U5Z0) USTEKINUMAB 90 mg in 1 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 76 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.04 mg in 1 mL HISTIDINE (UNII: 4QD397987E) 1 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-061-02 1 in 1 CARTON 09/25/2009 09/30/2009 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:57894-061-03 1 in 1 CARTON 09/25/2009 2 1 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC:57894-061-04 1 in 1 CARTON 09/25/2009 3 1 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125261 09/25/2009 STELARA
ustekinumab solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-054 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength USTEKINUMAB (UNII: FU77B4U5Z0) (USTEKINUMAB - UNII:FU77B4U5Z0) USTEKINUMAB 130 mg in 26 mL Inactive Ingredients Ingredient Name Strength EDETATE DISODIUM (UNII: 7FLD91C86K) 0.52 mg in 26 mL HISTIDINE (UNII: 4QD397987E) 20 mg in 26 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 27 mg in 26 mL METHIONINE (UNII: AE28F7PNPL) 10.4 mg in 26 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 10.4 mg in 26 mL SUCROSE (UNII: C151H8M554) 2210 mg in 26 mL Product Characteristics Color yellow (colorless to light yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-054-27 1 in 1 BOX 09/23/2016 1 26 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:57894-054-16 1 in 1 BOX 05/01/2017 2 26 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761044 09/23/2016 Labeler - Janssen Biotech, Inc. (099091753) Establishment Name Address ID/FEI Business Operations Cilag AG 483237103 manufacture(57894-060, 57894-061, 57894-054) , analysis(57894-060, 57894-061, 57894-054) Establishment Name Address ID/FEI Business Operations Janssen Biologics B.V. 409612918 analysis(57894-060, 57894-061, 57894-054) , api manufacture(57894-060, 57894-061, 57894-054) Establishment Name Address ID/FEI Business Operations Baxter Pharmaceutical Solutions 604719430 manufacture(57894-060, 57894-061) Establishment Name Address ID/FEI Business Operations Janssen Sciences Ireland UC 986030167 api manufacture(57894-060, 57894-061, 57894-054) , analysis(57894-060, 57894-061, 57894-054) Establishment Name Address ID/FEI Business Operations PPD Development Ireland Ltd. 985036175 analysis(57894-054)
