Label: VELTASSA- patiromer powder, for suspension




-
NDC Code(s):
53436-010-01,
53436-010-60,
53436-084-01,
53436-084-04, view more53436-084-30, 53436-084-91, 53436-084-92, 53436-168-01, 53436-168-30, 53436-252-01, 53436-252-30
- Packager: Vifor Pharma, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 2, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VELTASSA® safely and effectively. See full prescribing information for VELTASSA.
VELTASSA (patiromer) for oral suspension
Initial U.S. Approval: 2015INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- For adults, the recommended starting dose of Veltassa is 8.4 grams administered orally once daily. (2.2)
- For adults, adjust dose by 8.4 grams daily as needed at one-week intervals to obtain desired serum potassium target range. (2.2)
- For pediatric patients 12 to 17 years of age, the recommended starting dose of Veltassa is 4 grams administered orally once daily. (2.2)
- For pediatric patients 12 to 17 years of age, adjust dose by 4 grams daily as needed at one-week intervals to obtain desired serum potassium target range. (2.2)
- The maximum recommended dosage in adults and pediatric patients older than 12 years is 25.2 grams once daily. (2.2)
DOSAGE FORMS AND STRENGTHS
- Powder: 1 gram, 8.4 grams, 16.8 grams and 25.2 grams patiromer packets. (3)
CONTRAINDICATIONS
- Known hypersensitivity to Veltassa or any of its components. (4)
ADVERSE REACTIONS
- Most common adverse reactions (incidence ≥ 2%) are constipation, hypomagnesemia, diarrhea, nausea, abdominal discomfort and flatulence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Vifor Pharma, Inc. at 1-844-VELTASSA (1-844-835-8277) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Information
2.2 Recommended Dosing and Titration
2.3 Preparation of Veltassa
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Worsening of Gastrointestinal Motility
5.2 Hypomagnesemia
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Clinically Important Interaction of Veltassa with Other Drugs
7.2 No Observed Clinically Important Interaction of Veltassa with Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Two-Part, Randomized Withdrawal Study
14.2 One-Year Study
14.3 Pediatric Study
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Stability and Storage
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Veltassa is indicated for the treatment of hyperkalemia in adults and pediatric patients ages 12 years and older.
Limitation of Use: Veltassa should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action [see Clinical Pharmacology (12.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 General Information
Administer Veltassa at least 3 hours before or 3 hours after other oral medications except those shown to not have a clinically important interaction [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Do not heat Veltassa (e.g., microwave) or add to heated foods or liquids. Do not take Veltassa in its dry form.
2.2 Recommended Dosing and Titration
The recommended starting dose of Veltassa varies with age and is shown below. Multiple packets may be used to achieve the prescribed dose. Monitor serum potassium and adjust the dose of Veltassa based on the serum potassium level and the desired target range. The dose may be increased or decreased, as necessary, to reach the desired serum potassium concentration, up to a maximum dose of 25.2 grams once daily in adults and pediatric patients aged 12 years and older.
2.3 Preparation of Veltassa
Prepare each dose immediately prior to administration.
Measure 1/3 cup of water. Pour half of the water into a glass, then add the packet(s) of Veltassa and stir. Add the remaining half of the water and stir thoroughly. The powder will not dissolve and the mixture will look cloudy. Add more water to the mixture as needed for desired consistency.
Drink the mixture immediately. If powder remains in the glass after drinking, add more water, stir and drink immediately. Repeat as needed to ensure the entire dose is administered.
Other beverages or soft foods (e.g., apple sauce, yogurt, pudding) can be used instead of water to prepare the mixture by following the same steps as described above. A minimum volume of 45 mL (3 tablespoons) can be used to prepare doses up to and including 4 grams patiromer.
The potassium content of liquids or soft foods used to prepare the mixture should be considered as part of the dietary recommendations on potassium intake for each individual patient.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Veltassa is contraindicated in patients with a history of a hypersensitivity reaction to Veltassa or any of its components [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Worsening of Gastrointestinal Motility
Avoid use of Veltassa in patients with severe constipation, bowel obstruction or impaction, including abnormal post-operative bowel motility disorders, because Veltassa may be ineffective and may worsen gastrointestinal conditions.
Patients with a history of bowel obstruction or major gastrointestinal surgery, severe gastrointestinal disorders, or swallowing disorders were not included in the clinical studies.
5.2 Hypomagnesemia
Veltassa binds to magnesium in the colon, which can lead to hypomagnesemia. In clinical studies, hypomagnesemia was reported as an adverse reaction in 5.3% of adult patients treated with Veltassa [see Adverse Reactions (6.1)]. Monitor serum magnesium. Consider magnesium supplementation in patients who develop low serum magnesium levels on Veltassa.
-
6 ADVERSE REACTIONS
The following adverse reaction is discussed in greater detail elsewhere in the label:
- Hypomagnesemia [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Adult Patients
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of Veltassa cannot be directly compared to rates in the clinical trials of other drugs and may not reflect the rates observed in practice.
In the safety and efficacy clinical trials, 666 adult patients received at least one dose of Veltassa, including 219 exposed for at least 6 months and 149 exposed for at least one year.
Table 1 provides a summary of the most common adverse reactions (occurring in ≥ 2% of patients) in adult patients treated with Veltassa in these clinical trials. Most adverse reactions were mild to moderate. Constipation generally resolved during the course of treatment.
Table 1: Adverse Reactions Reported in ≥ 2% of Patients Adverse Reactions Adult Patients treated with Veltassa
(N=666)Note: Diarrhea is an aggregate term for Diarrhea and frequent bowel movements. Abdominal discomfort is an aggregate term for Abdominal discomfort, abdominal pain, abdominal pain upper, and abdominal pain lower. Constipation 7.2% Hypomagnesemia 5.3% Diarrhea 4.8% Nausea 2.3% Abdominal discomfort 2.0% Flatulence 2.0% The most commonly reported adverse reactions leading to discontinuation of Veltassa were gastrointestinal adverse reactions (2.7%), including vomiting (0.8%), diarrhea (0.6%), constipation (0.5%) and flatulence (0.5%).
Mild to moderate hypersensitivity reactions were reported in 0.3% of adult patients treated with Veltassa in clinical trials. Reactions have included edema of the lips.
-
7 DRUG INTERACTIONS
Veltassa has the potential to bind some oral co-administered medications, which could decrease their gastrointestinal absorption. Binding of Veltassa to other oral medications not listed in Table 3 below could cause decreased gastrointestinal absorption and loss of efficacy when taken close to the time Veltassa is administered. Administer other oral medications at least 3 hours before or 3 hours after Veltassa [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
7.1 Clinically Important Interaction of Veltassa with Other Drugs
The in-vitro binding of the following drugs to patiromer was evaluated and potentially clinically significant binding was observed. Some drugs were subsequently tested in-vivo and significant reduction in systemic exposure was observed [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Binding by Veltassa may reduce the systemic exposure and decrease the clinical efficacy of the co-administered drugs shown in Table 2. The administration of these drugs (and any drugs not listed in Table 3) should be separated by at least 3 hours from Veltassa.
Table 2: Clinically important drug interactions of Veltassa Angiotensin II receptor blockers (ARB)
Telmisartanβ-adrenoceptor blockers (β-blocker)
Bisoprolol, carvedilol, nebivololAntibiotics
CiprofloxacinAnti-Parathyroid Agents and Thyroid Preparations
LevothyroxineBlood Glucose Lowering Drugs
MetforminImmunosuppressants
Mycophenolate mofetilOthers
Quinidine, thiamine7.2 No Observed Clinically Important Interaction of Veltassa with Other Drugs
The binding of the following drugs to patiromer was evaluated [see Clinical Pharmacology (12.3)] and no clinically significant binding was observed. No separation of dosing is required for these drugs.
Table 3: No observed clinically important drug interactions of Veltassa Angiotensin-converting enzyme (ACE) inhibitors
Benazepril, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, ramipril, trandolaprilAngiotensin II receptor blockers (ARB)
Azilsartan, candesartan, irbesartan, losartan, olmesartan, valsartanβ-adrenoceptor blockers (β-blocker)
MetoprololLoop diuretics
Furosemide, bumetanide, torasemideMineralocorticoid receptor antagonists (MRA)
Eplerenone, finerenone, spironolactoneNeprilysin inhibitors
SacubitrilSodium-glucose cotransporter-2 (SGLT-2) inhibitors
Canagliflozin, dapagliflozin, empagliflozinAntibiotics
Trimethoprim, amoxicillin, cephalexinAnticoagulants
Warfarin, apixaban, rivaroxabanAnti-parathyroid agents and Thyroid preparations
CinacalcetAntithrombotic agents
Clopidogrel, acetylsalicylic acidBlood glucose lowering drugs
GlipizideCalcium channel blockers
Amlodipine, verapamilImmunosuppressants
TacrolimusOthers
Lithium, allopurinol, atorvastatin, digoxin, phenytoin, riboflavin, sevelamer -
8 USE IN SPECIFIC POPULATIONS
8.4 Pediatric Use
The safety and effectiveness of Veltassa for lowering serum potassium levels have been established in pediatric patients ages 12 years and older. Use of Veltassa for this indication is supported by evidence from an adequate and well-controlled study in adults, with additional pharmacodynamic and safety data in pediatric patients aged 12 years and older [see Dosage and Administration (2.2), Adverse Reactions (6.1), and Clinical Studies (14.3)].
Safety and efficacy have not been established in pediatric patients below the age of 12 years. Although the pediatric study included 9 patients 6 to less than 12 years of age, the dosing regimen that was evaluated in these patients did not appear to be effective in reducing serum potassium levels in this age group after 2 weeks. The starting dose of Veltassa in this age group was 2 g/day and the median dose at Day 14 was 6 g/day. In this age group the mean change in serum potassium from Baseline to Day 14 was -0.1 mEq/L (95% CI -0.7, 0.4). Because the available data are not sufficient to determine a safe and effective dosing regimen in patients 6 to less than 12 years of age, labeling recommendations cannot be provided for this age group.
8.5 Geriatric Use
Of the 666 patients treated with Veltassa in clinical studies, 60% were age 65 and over, and 20% were age 75 and over. No overall differences in effectiveness were observed between these patients and younger patients. Patients age 65 and older reported more gastrointestinal adverse reactions than younger patients.
8.6 Renal Impairment
Of the 666 adult patients treated with Veltassa in clinical studies, 93% had chronic kidney disease (CKD). All 14 pediatric patients ages 12 years and older treated with Veltassa in the clinical study had chronic kidney disease. No special dosing adjustments are needed for patients with renal impairment.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Veltassa is a powder for suspension in water for oral administration. The active ingredient is patiromer sorbitex calcium which consists of the active moiety, patiromer, a non-absorbed potassium-binding polymer, and a calcium-sorbitol counterion. Each gram of patiromer is equivalent to a nominal amount of 2 grams of patiromer sorbitex calcium.
The chemical name for patiromer sorbitex calcium is cross-linked polymer of calcium 2-fluoroprop-2-enoate with diethenylbenzene and octa-1,7-diene, combination with D-glucitol.
Patiromer sorbitex calcium is an amorphous, free-flowing powder that is composed of individual spherical beads. Patiromer sorbitex calcium is insoluble in solvents such as water, 0.1 M HCl, n-heptane and methanol. The chemical structure of patiromer sorbitex calcium is presented in Figure 1.
Figure 1: Chemical Structure of Patiromer Sorbitex Calcium
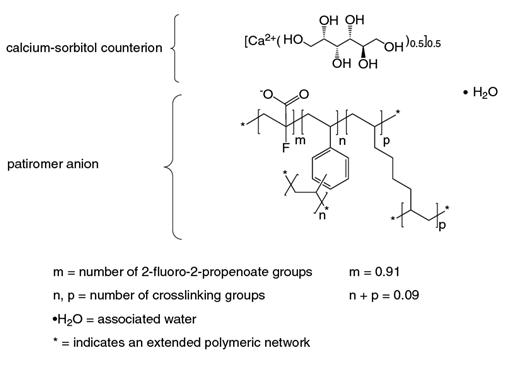
Each packet of Veltassa contains 1 gram, 8.4 grams, 16.8 grams or 25.2 grams of patiromer, the active moiety. The inactive ingredient is xanthan gum.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Veltassa is a non-absorbed, cation exchange polymer that contains a calcium-sorbitol counterion.
Veltassa increases fecal potassium excretion through binding of potassium in the lumen of the gastrointestinal tract. Binding of potassium reduces the concentration of free potassium in the gastrointestinal lumen, resulting in a reduction of serum potassium levels.
12.2 Pharmacodynamics
In a Phase 1 study in healthy adult subjects (6 to 8 subjects per group), Veltassa (0 grams to 50.4 grams per day) administered three times a day for 8 days caused a dose-dependent increase in fecal potassium excretion. A corresponding dose-dependent decrease in urinary potassium excretion with no change in serum potassium were also observed. Compared to placebo, Veltassa doses of 25.2 and 50.4 grams per day significantly decreased mean daily urinary potassium excretion.
In a Phase 1, open-label, multiple-dose crossover study in 12 healthy subjects, 25.2 grams of patiromer per day was administered orally as a once daily, twice daily or thrice daily regimen for 6 days in a randomly assigned order. A significant increase in mean daily fecal potassium excretion and concomitant decrease in mean daily urinary potassium excretion were observed during the treatment periods for all three dosing regimens. The mean increase in fecal potassium excretion ranged from 1283 to 1550 mg/day, and the mean decrease in urinary potassium excretion ranged from 1438 to 1534 mg/day across the three dosing regimens. No significant differences were observed among the dosing regimens with respect to mean daily fecal potassium and urinary potassium excretion. This was true for the overall comparison among the three dosing regimens, as well as for the pairwise comparisons.
In an open-label, uncontrolled study, 25 patients with hyperkalemia (mean baseline serum potassium of 5.9 mEq/L) and chronic kidney disease were given a controlled potassium diet for 3 days, followed by 16.8 grams patiromer daily (as divided doses) for 2 days while the controlled diet was continued. A statistically significant reduction in serum potassium (-0.2 mEq/L) was observed at 7 hours after the first dose. Serum potassium levels continued to decline during the 48-hour treatment period (-0.8 mEq/L at 48 hours after the first dose). Potassium levels remained stable for 24 hours after the last dose, then rose during the 4-day observation period following discontinuation of Veltassa.
12.3 Pharmacokinetics
Absorption
In radiolabeled ADME studies in rats and dogs, patiromer was not systemically absorbed and was excreted in the feces. Quantitative whole-body autoradiography analysis in rats demonstrated that radioactivity was limited to the gastrointestinal tract, with no detectable level of radioactivity in any other tissues or organs.
Effect of Food
Veltassa can be taken with or without food. In an open-label study, 114 patients with hyperkalemia were randomized to Veltassa once daily with food or without food. Serum potassium at the end of treatment, the change from baseline in serum potassium, and the mean dose of Veltassa were similar between groups.
Drug Interactions
Veltassa has the potential to bind some oral co-administered medications, which could decrease their gastrointestinal absorption.
Fifty-six (56) drugs were tested in-vitro to determine the potential for interaction with Veltassa [see Drug Interactions (7)]. For oral drug products not listed in Table 3, administration of patiromer should be separated by at least 3 hours as a precautionary measure.
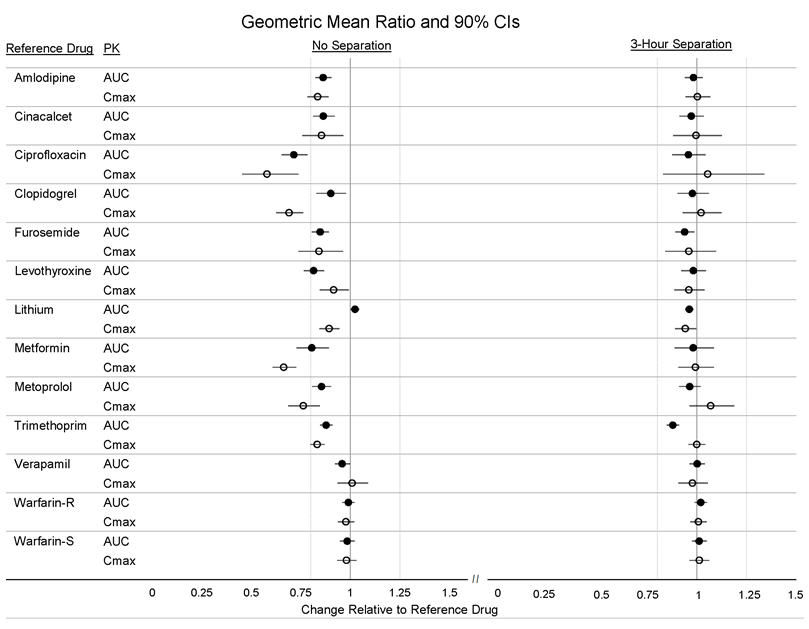
Twelve (12) drugs that showed an in vitro interaction were subsequently tested in vivo. These studies in healthy volunteers showed that Veltassa did not alter the systemic exposure of amlodipine, cinacalcet, clopidogrel, furosemide, lithium, metoprolol, trimethoprim, verapamil or warfarin when coadministered with Veltassa. Veltassa decreased the systemic exposure of coadministered ciprofloxacin, levothyroxine and metformin. However, there was no interaction when Veltassa and these drugs were taken 3 hours apart (Figure 2) [see Drug Interactions (7)].
Figure 2: Effects of Veltassa on the Pharmacokinetic Exposures of Other Orally Administered Medications with No Dosing Separation and with a 3-Hour Separation
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Patiromer was not genotoxic in the reverse mutation test (Ames assay), chromosome aberration or rat micronucleus assays.
Carcinogenicity studies have not been performed.
Patiromer did not impair the fertility in male or female rats at doses up to 10-fold the maximum recommended human dose (MRHD).
-
14 CLINICAL STUDIES
14.1 Two-Part, Randomized Withdrawal Study
The efficacy of Veltassa was demonstrated in a two-part, single-blind randomized withdrawal study that evaluated Veltassa in hyperkalemic patients with CKD on stable doses of at least one renin-angiotensin-aldosterone system inhibitor (i.e., angiotensin-converting enzyme inhibitor, angiotensin II receptor blocker, or aldosterone antagonist).
In Part A, 243 patients were treated with Veltassa for 4 weeks. Patients with a baseline serum potassium of 5.1 mEq/L to < 5.5 mEq/L received a starting Veltassa dose of 8.4 grams patiromer per day (as a divided dose) and patients with a baseline serum potassium of 5.5 mEq/L to < 6.5 mEq/L received a starting Veltassa dose of 16.8 grams patiromer per day (as a divided dose). The dose of Veltassa was titrated, as needed, based on the serum potassium level, assessed starting on Day 3 and then at weekly visits (Weeks 1, 2 and 3) to the end of the 4-week treatment period, with the aim of maintaining serum potassium in the target range (3.8 mEq/L to < 5.1 mEq/L).
The mean age of patients was 64 years, 58% of patients were men, and 98% were white. Approximately 97% of patients had hypertension, 57% had type 2 diabetes, and 42% had heart failure.
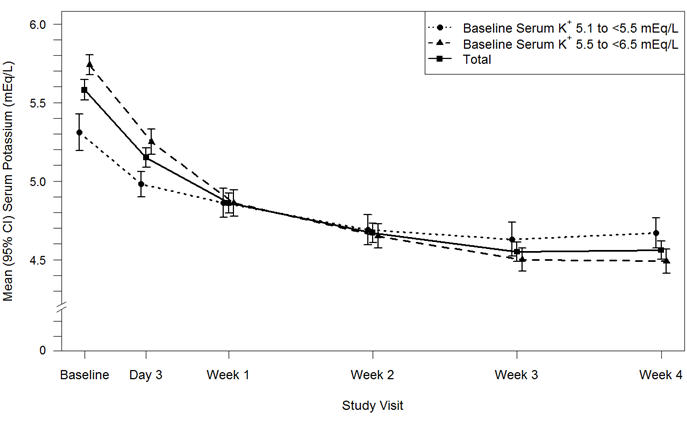
Results for the Part A primary endpoint, the change in serum potassium from Baseline to Week 4, are summarized in Table 4. Mean serum potassium over time for the intent-to-treat population is displayed in Figure 3. For the Part A secondary endpoint, 76% (95% CI: 70%, 81%) of patients had a serum potassium in the target range of 3.8 mEq/L to < 5.1 mEq/L at Week 4. The mean daily doses of Veltassa were 13 grams and 21 grams in patients with serum potassium of 5.1 to < 5.5 mEq/L and 5.5 to < 6.5 mEq/L, respectively.
Table 4: Veltassa Treatment Phase (Part A): Primary Endpoint Baseline, mean (SD) Week 4 Change from Baseline, Mean ± SE
(95% CI)p-value Baseline Potassium 5.1 to <5.5 mEq/L (n=90) Serum Potassium (mEq/L) 5.31 (0.57) -0.65 ± 0.05
(-0.74, -0.55)Baseline Potassium 5.5 to <6.5 mEq/L (n=147) 5.74 (0.40) -1.23 ± 0.04
(-1.31, -1.16)Overall Population (n=237) 5.58 (0.51) -1.01 ± 0.03
(-1.07, -0.95)< 0.001 Figure 3: Estimated Mean (95% CI) of Central Serum Potassium (mEq/L) Over Time
In Part B, 107 patients with a Part A baseline serum potassium of 5.5 mEq/L to < 6.5 mEq/L and whose serum potassium was in the target range (3.8 mEq/L to < 5.1 mEq/L) at Part A Week 4 and still receiving RAAS inhibitor medication were randomized to continue Veltassa or to receive placebo to evaluate the effect of withdrawing Veltassa on serum potassium. In patients randomized to Veltassa, the mean daily dose was 21 grams at the start of Part B and during Part B.
The Part B primary endpoint was the change in serum potassium from Part B baseline to the earliest visit at which the patient's serum potassium was first outside of the range of 3.8 to < 5.5 mEq/L, or to Part B Week 4 if the patient's serum potassium remained in the range. In Part B, serum potassium rose by 0.72 mEq/L in patients who were switched to placebo, versus no change in patients who remained on Veltassa. Results are summarized in Table 5.
Table 5: Randomized, Placebo-Controlled Withdrawal Phase (Part B): Primary Endpoint Placebo
(n=52)Veltassa
(n=55)Difference Estimate (95% CI) p-value Estimated Median Change in Serum Potassium from Baseline (mEq/L) 0.72 0.00 0.72
(0.46, 0.99)< 0.001 More placebo patients (91%; 95% CI: 83%, 99%) developed a serum potassium ≥ 5.1 mEq/L at any time during Part B than Veltassa patients (43%; 95% CI: 30%, 56%), p < 0.001. More placebo patients (60%; 95% CI: 47%, 74%) developed a serum potassium ≥ 5.5 mEq/L at any time during Part B than Veltassa patients (15%; 95% CI: 6%, 24%), p < 0.001.
14.2 One-Year Study
The effect of treatment with Veltassa for up to 52 weeks was evaluated in an open-label study of 304 hyperkalemic patients with CKD and type 2 diabetes mellitus on RAAS inhibitor therapy. Figure 4 shows that the treatment effect on serum potassium was maintained during continued therapy. In patients with a baseline serum potassium of > 5.0 to 5.5 mEq/L who received an initial dose of 8.4 grams patiromer per day (as a divided dose), the mean daily dose was 14 grams; in those with a baseline serum potassium of > 5.5 to < 6.0 mEq/L who received an initial dose of 16.8 grams patiromer per day (as a divided dose), the mean daily dose was 20 grams during the entire study.
Figure 4: Mean (95% CI) Serum Potassium over Time
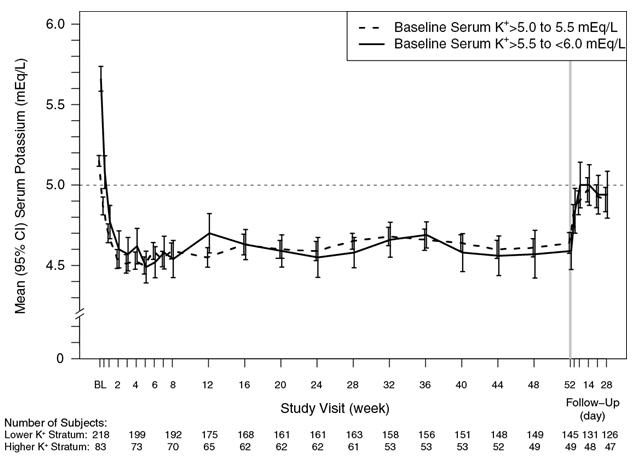
14.3 Pediatric Study
The potassium-lowering effect of Veltassa was evaluated in an open-label, single-arm study in pediatric patients 12 to 17 years of age with CKD and hyperkalemia. The study included an initial 14-day dose finding phase, followed by an up to 24-week long-term (LT) treatment phase and a 2-week follow-up period. Veltassa was given once daily as a powder for oral suspension. The dose of Veltassa was titrated, as needed, based on the serum potassium level, assessed starting on Day 3 and then at Day 7 and Day 14, with the aim of maintaining serum potassium in the target range (3.8 mEq/L to < 5.0 mEq/L).
All 14 patients 12 to 17 years of age completed the dose finding phase. The mean age was 14.5 years, 79% were male, and all were white. The average weight at baseline was 51 kg and 57% of patients had a baseline eGFR <30 mL/min/1.73 m2. Approximately 57% were on RAAS inhibitor therapy at baseline. The mean (SD) baseline serum potassium was 5.5 mEq/L (0.3 mEq/L).
The mean change in serum potassium from baseline to Day 14 was -0.5 mEq/L (95% CI -0.8, -0.2). The proportion of patients 12 to 17 years of age with a serum potassium within the normal range was 50% at Day 14. The median dose at Day 14 was 4.2 g/day.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Veltassa is supplied as a powder for oral suspension formulated with xanthan gum. Veltassa is packaged in single-use packets containing 1 gram, 8.4 grams, 16.8 grams or 25.2 grams patiromer as follows:
Veltassa (grams) Carton of 4 Packets Carton of 30 Packets Carton of 60 Packets 1 - - NDC 53436-010-60 8.4 NDC 53436-084-04 NDC 53436-084-30 - 16.8 - NDC 53436-168-30 - 25.2 - NDC 53436-252-30 - 16.2 Stability and Storage
Veltassa should be stored in the refrigerator at 2°C to 8°C (36°F to 46°F).
If stored at room temperature (25°C ± 2°C [77°F ± 4°F]), Veltassa must be used within 3 months of being taken out of the refrigerator. For either storage condition, do not use Veltassa after the expiration date printed on the packet.
Avoid exposure to excessive heat above 40°C (104°F).
-
17 PATIENT COUNSELING INFORMATION
Drug Interactions
Advise patients who are taking other oral medication that separation of dosing of Veltassa by at least 3 hours (before or after) may be needed except those shown to not have a clinically important interaction in Table 3 above [see Drug Interactions (7)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 8.4 g Packet Carton
- PRINCIPAL DISPLAY PANEL - 16.8 g Packet Carton
- PRINCIPAL DISPLAY PANEL - 25.2 g Packet Carton
- PRINCIPAL DISPLAY PANEL - 1 g Packet Carton
-
INGREDIENTS AND APPEARANCE
VELTASSA
patiromer powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53436-084 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength patiromer (UNII: 1FQ2RY5YHH) (patiromer - UNII:1FQ2RY5YHH) patiromer 8.4 g Inactive Ingredients Ingredient Name Strength xanthan gum (UNII: TTV12P4NEE) 0.12 g Product Characteristics Color WHITE (off-white to light-brown) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53436-084-04 4 in 1 CARTON 10/23/2015 1 NDC:53436-084-01 1 in 1 PACKET; Type 0: Not a Combination Product 2 NDC:53436-084-30 30 in 1 CARTON 10/23/2015 2 NDC:53436-084-01 1 in 1 PACKET; Type 0: Not a Combination Product 3 NDC:53436-084-92 4 in 1 CARTON 05/01/2016 3 NDC:53436-084-91 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205739 10/23/2015 VELTASSA
patiromer powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53436-168 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength patiromer (UNII: 1FQ2RY5YHH) (patiromer - UNII:1FQ2RY5YHH) patiromer 16.8 g Inactive Ingredients Ingredient Name Strength xanthan gum (UNII: TTV12P4NEE) 0.24 g Product Characteristics Color WHITE (off-white to light-brown) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53436-168-30 30 in 1 CARTON 10/23/2015 1 NDC:53436-168-01 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205739 10/23/2015 VELTASSA
patiromer powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53436-252 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength patiromer (UNII: 1FQ2RY5YHH) (patiromer - UNII:1FQ2RY5YHH) patiromer 25.2 g Inactive Ingredients Ingredient Name Strength xanthan gum (UNII: TTV12P4NEE) 0.36 g Product Characteristics Color WHITE (off-white to light-brown) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53436-252-30 30 in 1 CARTON 10/23/2015 1 NDC:53436-252-01 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205739 10/23/2015 VELTASSA
patiromer powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53436-010 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength patiromer (UNII: 1FQ2RY5YHH) (patiromer - UNII:1FQ2RY5YHH) patiromer 1 g Inactive Ingredients Ingredient Name Strength xanthan gum (UNII: TTV12P4NEE) 0.014 g Product Characteristics Color WHITE (off-white to light-brown) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53436-010-60 60 in 1 CARTON 10/02/2024 1 NDC:53436-010-01 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205739 10/02/2024 Labeler - Vifor Pharma, Inc. (808446087)